Email (for orders and customer service enquiries): cs-books@wiley.co.uk Visit ou r Home Page on www wileyeu rope com or www wiley com
All Rights Reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, mechanical, photocopying, recording, scanning or otherwise, except under the terms of the Copyright, Designs and Patents Act 1988 or under the terms of a licence issued by the Copyright Licensing Agency Ltd, 90 Tottenham Court Road, London W1T 4LP, UK, without the permission in writing of the Publisher. Requests to the Publisher should be addressed to the Permissions Department, John Wiley & Sons Ltd, The Atrium, Southern Gate, Chichester, West Sussex PO19 8SQ, England, or emailed to permreq@wiley.co.uk, or faxed to ( 44) 1243 770620.
Designations used by companies to distinguish their products are often claimed as trademarks. All brand names and product names used in this book are trade names, service marks, trademarks or registered trademarks of their respective owners. The Publisher is not associated with any product or vendor mentioned in this book.
This publication is designed to provide accurate and authoritative information in regard to the subject matter covered. It is sold on the understanding that the Publisher is not engaged in rendering professional services. If professional advice or other expert assistance is required, the services of a competent professional should be sought.
Other Wiley Editorial Offi ces
John Wiley & Sons Inc., 111 River Street, Hoboken, NJ 07030, USA
Jossey-Bass, 989 Market Street, San Francisco, CA 94103-1741, USA
A catalogue record for this book is available from the British Library
ISBN 978-0-470-01244-4 (HB)
ISBN 978-0-470-01245-1 (PB)
Typeset in 10.5/12.5 pt Times by Thomson Digital
Printed and bound in Great Britain by Antony Rowe Ltd., Chippenham, Wilts
This book is printed on acid-free paper responsibly manufactured from sustainable forestry in which at least two trees are planted for each one used for paper production.
I dedicate this book to my beautiful daughter Alice. To borrow a phrase: ‘without her help, it would have been written in half the time’!
13.3.8 Additional therapeutic applications of monoclonal antibodies
13.4 Vaccine technology
13.4.1 Traditional vaccine preparations
13.4.1.1 Attenuated, dead or inactivated bacteria
13.4.1.2 Attenuated and inactivated viral vaccines
13.4.1.3 Toxoids and antigen-based vaccines
13.4.2 The impact of genetic engineering on vaccine technology
13.4.3 Peptide vaccines
13.4.4 Vaccine vectors
13.4.5 Development of an AIDS vaccine
13.5
13.5.3 Oil-based emulsion adjuvants
13.5.4 Bacteria/bacterial
14.3.4
14.4
14.5
14.6
14.7.1
14.7.2
14.8
Preface
This book has been written as a sister publication to Biopharmaceuticals: Biochemistry and Biotechnology, a second edition of which was published by John Wiley and Sons in 2003. The latter textbook caters mainly for advanced undergraduate/postgraduate students undertaking degree programmes in biochemistry, biotechnology and related disciplines. Such students have invariably pursued courses/modules in basic protein science and molecular biology in the earlier parts of their degree programmes; hence, the basic principles of protein structure and molecular biology were not considered as part of that publication. This current publication is specifically tailored to meet the needs of a broader audience, particularly to include students undertaking programmes in pharmacy/pharmaceutical science, medicine and other branches of biomedical/clinical sciences. Although evolving from Biopharmaceuticals: Biochemistry and Biotechnology, its focus is somewhat different, reflecting its broader intended readership. This text, therefore, includes chapters detailing the basic principles of protein structure and molecular biology. It also increases/extends the focus upon topics such as formulation and delivery of biopharmaceuticals, and it contains numerous case studies in which both biotech and clinical aspects of a particular approved product of pharmaceutical biotechnology are overviewed. The book, of course, should also meet the needs of students undertaking programmes in core biochemistry, biotechnology or related scientific areas and be of use as a broad reference source to those already working within the pharmaceutical biotechnology sector.
As always, I owe a debt of gratitude to the various people who assisted in the completion of this textbook. Thanks to Sandy for her help in preparing various figures, usually at ridiculously short notice. To Gerard Wall, for all the laughs and for several useful discussions relating to molecular biology. Thank you to Nancy, my beautiful wife, for accepting my urge to write (rather than to change baby’s nappies) with good humour – most of the time anyway! I am also grateful to the staff of John Wiley and Sons for their continued professionalism and patience with me when I keep overrunning submission deadlines. Finally, I have a general word of appreciation to all my colleagues at the University of Limerick for making this such an enjoyable place to work.
Gary Walsh November 2006
Acronyms
ADCC antibody-dependent cell cytoxicity
BAC bacterial artificial chromosome
BHK baby hamster kidney
cDNA complementary DNA
CHO Chinese hamster ovary
CNTF ciliary neurotrophic factor
CSF colony-stimulating factor
dsRNA double-stranded RNA
EDTA ethylenediaminetetraacetic acid
ELISA enzyme-linked immunosorbent assay
EPO erythropoietin
FGF fibroblast growth factor
FSH follicle-stimulating hormone
GDNF glial cell-derived neurotrophic factor
GH growth hormone
hCG human chorionic gonadotrophin
HIV human immunodeficiency virus
HPLC high-performance liquid chromatography
IGF insulin-like growth factor
ISRE interferon-stimulated response element
JAK Janus kinase
LAF lymphocyte activating factor
LIF leukaemia inhibitory factor
LPS lipopolysaccharide
MHC major histocompatibility complex
MPS mucopolysaccharidosis
mRNA messenger RNA
PDGF platelet-derived growth factor
PEG polyethylene glycol
PTK protein tyrosine kinase
PTM post-translational modification
rDNA recombinant DNA
RNAi RNA interference
rRNA ribosomal RNA
SDS sodium dodecyl sulfate
ssRNA single-stranded RNA
STATs signal transducers and activators of transcription
TNF tumour necrosis factor
tPA tissue plasminogen activator
tRNA transfer RNA
WAP whey acid protein
WFI water for injections
1 Pharmaceuticals, biologics and biopharmaceuticals
1.1 Introduction to pharmaceutical products
Pharmaceutical substances form the backbone of modern medicinal therapy. Most traditional pharmaceuticals are low molecular weight organic chemicals (Table 1.1). Although some (e.g. aspirin) were originally isolated from biological sources, most are now manufactured by direct chemical synthesis. Two types of manufacturing company thus comprise the ‘traditional’ pharmaceutical sector: the chemical synthesis plants, which manufacture the raw chemical ingredients in bulk quantities, and the finished product pharmaceutical facilities, which purchase these raw bulk ingredients, formulate them into final pharmaceutical products, and supply these products to the end user.
In addition to chemical-based drugs, a range of pharmaceutical substances (e.g. hormones and blood products) are produced by/extracted from biological sources. Such products, some major examples of which are listed in Table 1.2, may thus be described as products of biotechnology. In some instances, categorizing pharmaceuticals as products of biotechnology or chemical synthesis becomes somewhat artificial. For example, certain semi-synthetic antibiotics are produced by chemical modification of natural antibiotics produced by fermentation technology.
1.2 Biopharmaceuticals and pharmaceutical biotechnology
Terms such as ‘biologic’, ‘biopharmaceutical’ and ‘products of pharmaceutical biotechnology’ or ‘biotechnology medicines’ have now become an accepted part of the pharmaceutical literature. However, these terms are sometimes used interchangeably and can mean different things to different people. Although it might be assumed that ‘biologic’ refers to any pharmaceutical product produced by biotechnological endeavour, its definition is more limited. In pharmaceutical circles, ‘biologic’ generally refers to medicinal products derived from blood, as well as vaccines, toxins and allergen products. ‘Biotechnology’ has a much broader and long-established meaning. Essentially, it refers
Table 1.1 Some traditional pharmaceutical substances that are generally produced by direct chemical synthesis
to the use of biological systems (e.g. cells or tissues) or biological molecules (e.g. enzymes or antibodies) for/in the manufacture of commercial products.
The term ‘biopharmaceutical’ was first used in the 1980s and came to describe a class of therapeutic proteins produced by modern biotechnological techniques, specifically via genetic engineering (Chapter 3) or, in the case of monoclonal antibodies, by hybridoma technology (Chapter 13). Although the majority of biopharmaceuticals or biotechnology products now approved or in development are proteins produced via genetic engineering, these terms now also encompass nucleic-acid-based, i.e. deoxyribonucleic acid (DNA)- or ribonucleic acid (RNA)-based products, and whole-cell-based products.
1.3 History of the pharmaceutical industry
The pharmaceutical industry, as we now know it, is barely 60 years old. From very modest beginnings, it has grown rapidly, reaching an estimated value of US$100 billion by the mid 1980s. Its current value is likely double or more this figure. There are well in excess of 10 000 pharmaceutical companies in existence, although only about 100 of these can claim to be of true international significance. These companies manufacture in excess of 5000 individual pharmaceutical substances used routinely in medicine.
Table 1.2 Some pharmaceuticals that were traditionally obtained by direct extraction from biological source material. Many of the protein-based pharmaceuticals mentioned are now also produced by genetic engineering
SubstanceMedical application
Blood products (e.g. coagulation factors)Treatment of blood disorders such as haemophilia A or B
VaccinesVaccination against various diseases
AntibodiesPassive immunization against various diseases
InsulinTreatment of diabetes mellitus
EnzymesThrombolytic agents, digestive aids, debriding agents (i.e. cleansing of wounds)
AntibioticsTreatment against various infections agents
Plant extracts (e.g. alkaloids)Various, including pain relief
The fi rst stages of development of the modern pharmaceutical industry can be traced back to the turn of the twentieth century. At that time (apart from folk cures), the medical community had at their disposal only four drugs that were effective in treating specific diseases:
Digitalis (extracted from foxglove) was known to stimulate heart muscle and, hence, was used to treat various heart conditions.
Quinine, obtained from the barks/roots of a plant (Cinchona genus), was used to treat malaria.
Pecacuanha (active ingredient is a mixture of alkaloids), used for treating dysentery, was obtained from the bark/roots of the plant genus Cephaelis.
Mercury, for the treatment of syphilis.
This lack of appropriate, safe and effective medicines contributed in no small way to the low life expectancy characteristic of those times.
Developments in biology (particularly the growing realization of the microbiological basis of many diseases), as well as a developing appreciation of the principles of organic chemistry, helped underpin future innovation in the fledgling pharmaceutical industry. The successful synthesis of various artificial dyes, which proved to be therapeutically useful, led to the formation of pharmaceutical/chemical companies such as Bayer and Hoechst in the late 1800s. Scientists at Bayer, for example, succeeded in synthesizing aspirin in 1895.
Despite these early advances, it was not until the 1930s that the pharmaceutical industry began to develop in earnest. The initial landmark discovery of this era was probably the discovery, and chemical synthesis, of the sulfa drugs. These are a group of related molecules derived from the red dye prontosil rubrum . These drugs proved effective in the treatment of a wide variety of bacterial infections (Figure 1.1). Although it was first used therapeutically in the early 1920s, large-scale industrial production of insulin also commenced in the 1930s.
The medical success of these drugs gave new emphasis to the pharmaceutical industry, which was boosted further by the commencement of industrial-scale penicillin manufacture in the early 1940s. Around this time, many of the current leading pharmaceutical companies (or their forerunners) were founded. Examples include Ciba Geigy, Eli Lilly, Wellcome, Glaxo and Roche. Over the next two to three decades, these companies developed drugs such as tetracyclines, corticosteroids, oral contraceptives, antidepressants and many more. Most of these pharmaceutical substances are manufactured by direct chemical synthesis.
1.4 The age of biopharmaceuticals
Biomedical research continues to broaden our understanding of the molecular mechanisms underlining both health and disease. Research undertaken since the 1950s has pinpointed a host of proteins produced naturally in the body that have obvious therapeutic applications. Examples include the interferons and interleukins (which regulate the immune response), growth factors, such as erythropoietin (EPO; which stimulates red blood cell production), and neurotrophic factors (which regulate the development and maintenance of neural tissue).
Figure 1.1 Sulfa drugs and their mode of action. The first sulfa drug to be used medically was the red dye prontosil rubrum (a). In the early 1930s, experiments illustrated that the administration of this dye to mice infected with haemolytic streptococci prevented the death of the mice. This drug, although effective in vivo, was devoid of in vitro antibacterial activity. It was first used clinically in 1935 under the name Streptozon. It was subsequently shown that prontosil rubrum was enzymatically reduced by the liver, forming sulfanilamide, the actual active antimicrobial agent (b). Sulfanilamide induces its effect by acting as an anti-metabolite with respect to para -aminobenzoic acid (PABA) (c). PABA is an essential component of tetrahydrofolic acid (THF) (d). THF serves as an essential cofactor for several cellular enzymes. Sulfanilamide (at sufficiently high concentrations) inhibits manufacture of THF by competing with PABA. This effectively inhibits essential THF-dependent enzyme reactions within the cell. Unlike humans, who can derive folates from their diets, most bacteria must synthesize it de novo, as they cannot absorb it intact from their surroundings
Although the pharmaceutical potential of these regulatory molecules was generally appreciated, their widespread medical application was in most cases rendered impractical due to the tiny quantities in which they were naturally produced. The advent of recombinant DNA technology (genetic engineering) and monoclonal antibody technology (hybridoma technology) overcame many such difficulties, and marked the beginning of a new era of the pharmaceutical sciences.
Recombinant DNA technology has had a fourfold positive impact upon the production of pharmaceutically important proteins:
Prontosil rubrum (a) Sulphanilamide (b) PABA (c)
Pteridine derivative PABA Glutamic acid
Tetrahydrofolic acid (d)
It overcomes the problem of source availability. Many proteins of therapeutic potential are produced naturally in the body in minute quantities. Examples include interferons (Chapter 8), interleukins (Chapter 9) and colony-stimulating factors (CSFs; Chapter 10). This rendered impractical their direct extraction from native source material in quantities sufficient to meet likely clinical demand. Recombinant production (Chapters 3 and 5) allows the manufacture of any protein in whatever quantity it is required.
It overcomes problems of product safety. Direct extraction of product from some native biological sources has, in the past, led to the unwitting transmission of disease. Examples include the transmission of blood-borne pathogens such as hepatitis B and C and human immunodeficiency virus (HIV) via infected blood products and the transmission of Creutzfeldt–Jakob disease to persons receiving human growth hormone (GH) preparations derived from human pituitaries.
It provides an alternative to direct extraction from inappropriate/dangerous source material. A number of therapeutic proteins have traditionally been extracted from human urine. Folliclestimulating hormone (FSH), the fertility hormone, for example, is obtained from the urine of postmenopausal women, and a related hormone, human chorionic gonadotrophin (hCG), is extracted from the urine of pregnant women (Chapter 11). Urine is not considered a particularly desirable source of pharmaceutical products. Although several products obtained from this source remain on the market, recombinant forms have now also been approved. Other potential biopharmaceuticals are produced naturally in downright dangerous sources. Ancrod, for example, is a protein displaying anti-coagulant activity (Chapter 12) and, hence, is of potential clinical use. It is, however, produced naturally by the Malaysian pit viper. Although retrieval by milking snake venom is possible, and indeed may be quite an exciting procedure, recombinant production in less dangerous organisms, such as Escherichia coli or Saccharomycese cerevisiae, would be considered preferable by most.
It facilitates the generation of engineered therapeutic proteins displaying some clinical advantage over the native protein product. Techniques such as site-directed mutagenesis facilitate the logical introduction of predefined changes in a protein’s amino acid sequence. Such changes can be as minimal as the insertion, deletion or alteration of a single amino acid residue, or can be more substantial (e.g. the alteration/deletion of an entire domain, or the generation of a novel hybrid protein). Such changes can be made for a number of reasons, and several engineered products have now gained marketing approval. An overview summary of some engineered product types now on the market is provided in Table 1.3. These and other examples will be discussed in subsequent chapters.
Despite the undoubted advantages of recombinant production, it remains the case that many protein-based products extracted directly from native source material remain on the market. In certain circumstances, direct extraction of native source material can prove equally/more attractive than recombinant production. This may be for an economic reason if, for example, the protein is produced in very large quantities by the native source and is easy to extract/purify, e.g. human serum albumin (HSA; Chapter 12). Also, some blood factor preparations purified from donor blood actually contain several different blood factors and, hence, can be used to treat several haemophilia patient types. Recombinant blood factor preparations, on the other hand, contain but a single blood factor and, hence, can be used to treat only one haemophilia type (Chapter 12).
The advent of genetic engineering and monoclonal antibody technology underpinned the establishment of literally hundreds of start-up biopharmaceutical (biotechnology) companies in
Table 1.3 Selected engineered biopharmaceutical types/products that have now gained marketing approval. These and additional such products will be discussed in detail in subsequent chapters
Replacement of most/virtually all of the murine amino acid sequences with sequences found in human antibodies
‘Ontak’, a fusion protein (Chapter 9)Fusion protein consisting of the diphtheria toxin linked to interleukin-2 (IL-2)
Generation of a faster acting thrombolytic (clot degrading) agent
Production of a lower molecular mass product
Greatly reduced/eliminated immunogenicity. Ability to activate human effector functions
Targets toxin selectively to cells expressing an IL-2 receptor
the late 1970s and early 1980s. The bulk of these companies were founded in the USA, with smaller numbers of start-ups emanating from Europe and other world regions.
Many of these fledgling companies were founded by academics/technical experts who sought to take commercial advantage of developments in the biotechnological arena. These companies were largely financed by speculative monies attracted by the hype associated with the establishment of the modern biotech era. Although most of these early companies displayed significant technical expertise, the vast majority lacked experience in the practicalities of the drug development process (Chapter 4). Most of the well-established large pharmaceutical companies, on the other hand, were slow to invest heavily in biotech research and development. However, as the actual and potential therapeutic significance of biopharmaceuticals became evident, many of these companies did diversify into this area. Most either purchased small, established biopharmaceutical concerns or formed strategic alliances with them. An example was the long-term alliance formed by Genentech (see later) and the well-
Table 1.4 Pharmaceutical companies who manufacture and/or market biopharmaceutical products approved for general medical use in the USA and EU
Sanofi-AventisHoechst AG BayerWyeth
Novo NordiskGenzyme Isis PharmaceuticalsAbbott GenentechRoche CentocorNovartis
Boehringer ManheimSerono Galenus ManheimOrganon
Eli LillyAmgen
Ortho BiotechGlaxoSmithKline
Schering PloughCytogen
Hoffman-la-RocheImmunomedics ChironBiogen
established pharmaceutical company Eli Lilly. Genentech developed recombinant human insulin, which was then marketed by Eli Lilly under the trade name Humulin. The merger of biotech capability with pharmaceutical experience helped accelerate development of the biopharmaceutical sector. Many of the earlier biopharmaceutical companies no longer exist. The overall level of speculative finance available was not sufficient to sustain them all long term (it can take 6–10 years and US$800 million to develop a single drug; Chapter 4). Furthermore, the promise and hype of biotechnology sometimes exceeded its ability actually to deliver a final product. Some biopharmaceutical substances showed little efficacy in treating their target condition, and/or exhibited unacceptable side effects. Mergers and acquisitions also led to the disappearance of several biopharmaceutical concerns. Table 1.4 lists many of the major pharmaceutical concerns which now manufacture/market biopharmaceuticals approved for general medical use. Box 1.1 provides a profile of three well-established dedicated biopharmaceutical companies.
Box 1.1
Amgen, Biogen and Genentech
Amgen, Biogen and Genentech represent three pioneering biopharmaceutical companies that still remain in business.
Founded in the 1980s as AMGen (Applied Molecular Genetics), Amgen now employs over 9000 people worldwide, making it one of the largest dedicated biotechnology companies in existence. Its headquarters are situated in Thousand Oaks, California, although it has research, manufacturing, distribution and sales facilities worldwide. Company activities focus upon developing novel (mainly protein) therapeutics for application in oncology, inflammation, bone disease, neurology, metabolism and nephrology. By mid 2006, seven of its recombinant products had been approved for general medical use (the EPO-based products ‘Aranesp’ and ‘Epogen’ (Chapter 10), the CSF-based products ‘Neupogen’ and ‘Neulasta’ (Chapter 10), as well as the interleukin-1 (IL-1) receptor antagonist ‘Kineret’, the anti-rheumatoid arthritis fusion protein Enbrel (Chapter 9) and the keratinocyte growth factor ‘Kepivance’, indicated for the treatment of severe oral mucositis. Total product sales for 2004 reached US$9.9 billion. In July 2002, Amgen acquired Immunex Corporation, another dedicated biopharmaceutical company founded in Seattle in the early 1980s.
Biogen was founded in Geneva, Switzerland, in 1978 by a group of leading molecular biologists. Currently, its global headquarters are located in Cambridge, MA, and it employs in excess of 2000 people worldwide. The company developed and directly markets the interferonbased product ‘Avonex’ (Chapter 8), but also generates revenues from sales of other Biogendiscovered products that are licensed to various other pharmaceutical companies. These include Schering Plough’s ‘Intron A’ (Chapter 8) and a number of hepatitis B-based vaccines sold by SmithKline Beecham (SKB) and Merck (Chapter 13).
Genentech was founded in 1976 by scientist Herbert Boyer and the venture capitalist Robert Swanson. Headquartered in San Francisco, it employs almost 5000 staff worldwide and has 10 protein-based products on the market. These include hGHs (Nutropin, Chapter 11), the antibody-based products ‘Herceptin’ and ‘Rituxan’ (Chapter 13) and the thrombolytic agents ‘Activase’ and ‘TNKase’ (Chapter 12). The company also has 20 or so products in clinical trials. In 2004, it generated some US$4.6 billion in revenues.
1.5 Biopharmaceuticals: current status and future prospects
Approximately one in every four new drugs now coming on the market is a biopharmaceutical. By mid 2006, some 160 biopharmaceutical products had gained marketing approval in the USA and/or EU. Collectively, these represent a global biopharmaceutical market in the region of US$35 billion (Table 1.5), and the market value is estimated to surpass US$50 billion by 2010. The products include a range of hormones, blood factors and thrombolytic agents, as well as vaccines and monoclonal antibodies (Table 1.6). All but two are protein-based therapeutic agents. The exceptions are two nucleic-acid-based products: ‘Vitravene’, an antisense oligonucleotide, and ‘Macugen’, an aptamer (Chapter 14). Many additional nucleic-acid-based products for use in gene therapy or antisense technology are in clinical trials, although the range of technical difficulties that still beset this class of therapeutics will ensure that protein-based products will overwhelmingly predominate for the foreseeable future (Chapter 14).
Many of the initial biopharmaceuticals approved were simple replacement proteins (e.g. blood factors and human insulin). The ability to alter the amino acid sequence of a protein logically coupled to an increased understanding of the relationship between protein structure and function (Chapters 2 and 3) has facilitated the more recent introduction of several engineered therapeutic proteins (Table 1.3). Thus far, the vast majority of approved recombinant proteins have been produced in the bacterium E. coli, the yeast S. cerevisiae or in animal cell lines (most notably Chinese hamster ovary (CHO) cells or baby hamster kidney (BHK) cells. These production systems are discussed in Chapter 5.
Although most biopharmaceuticals approved to date are intended for human use, a number of products destined for veterinary application have also come on the market. One early such example is that of recombinant bovine GH (Somatotrophin), which was approved in the USA in the early 1990s and used to increase milk yields from dairy cattle. Additional examples of approved veterinary biopharmaceuticals include a range of recombinant vaccines and an interferon-based product (Table 1.7).
Table 1.5 Approximate annual market values of some leading approved biopharmaceutical products. Data gathered from various sources, including company home pages, annual reports and industry reports
Product (Company)Product description (use)Annual sales value (US$, billions)
Procrit (Amgen/Johnson & Johnson) EPO (treatment of anaemia)4.0
Epogen & Aranesp combined (Amgen) EPO (treatment of anaemia)4.0
Intron A (Schering Plough)IFN- α (treatment of leukaemia)0.3
Remicade (Johnson & Johnson)Monoclonal antibody based (treatment of Crohn’s disease)
Avonex (Biogen)Interferon-β (IFN-β ; treatment of multiple sclerosis)
Embrel (Wyeth)Monoclonal antibody based (treatment of rheumatoid arthritis)
Rituxan (Genentech)Monoclonal antibody based (nonHodgkin’s lymphoma)
Humulin (Eli Lilly)Insulin (diabetes)1.0
Table 1.6 Summary categorization of biopharmaceuticals approved for general medical use in the EU and/or USA by 2006
At least 1000 potential biopharmaceuticals are currently being evaluated in clinical trials, although the majority of these are in early stage trials. Vaccines and monoclonal antibody-based products represent the two biggest product categories. Regulatory factors (e.g. hormones and
Table 1.7 Some recombinant (r) biopharmaceuticals recently approved for veterinary application in the EU ProductCompanyIndication
VirbacReduction of mortality/clinical symptoms associated with canine parvovirus
Fort Dodge LaboratoriesImmunization of cats against various feline pathogens
IntervetActive immunization of sows
IntervetReduction in clinical signs of progressive atrophic rhinitis in piglets
IntervetImmunization of pigs against classical swine fever
IntervetImmunization of pigs against classical swine fever
cytokines) and gene therapy and antisense-based products also represent significant groupings. Although most protein-based products likely to gain marketing approval over the next 2–3 years will be produced in engineered E. coli, S. cerevisiae or animal cell lines, some products now in clinical trials are being produced in the milk of transgenic animals (Chapter 5). Additionally, plant-based transgenic expression systems may potentially come to the fore, particularly for the production of oral vaccines (Chapter 5).
Interestingly, the fi rst generic biopharmaceuticals are already entering the market. Patent protection for many fi rst-generation biopharmaceuticals (including recombinant human GH (rhGH), insulin, EPO, interferon- α (IFN- α) and granulocyte-CSF (G-CSF)) has now/is now coming to an end. Most of these drugs command an overall annual market value in excess of US$1 billion, rendering them attractive potential products for many biotechnology/pharmaceutical companies. Companies already/soon producing generic biopharmaceuticals include Biopartners (Switzerland), Genemedix (UK), Sicor and Ivax (USA), Congene and Microbix (Canada) and BioGenerix (Germany). Genemedix, for example, secured approval for sale of a recombinant CSF in China in 2001 and is also commencing the manufacture of recombinant EPO. Sicor currently markets hGH and IFN- α in eastern Europe and various developing nations. A generic hGH also gained approval in both Europe and the USA in 2006.
To date (mid 2006), no gene-therapy-based product has thus far been approved for general medical use in the EU or USA, although one such product (‘Gendicine’; Chapter 14) has been approved in China. Although gene therapy trials were initiated as far back as 1989, the results have been disappointing. Many technical difficulties remain in relation to, for example, gene delivery and regulation of expression. Product effectiveness was not apparent in the majority of trials undertaken and safety concerns have been raised in several trials.
Only one antisense-based product has been approved to date (in 1998) and, although several such antisense agents continue to be clinically evaluated, it is unlikely that a large number of such products will be approved over the next 3–4 years. Aptamers represent an additional emerging class of nucleic-acid-based therapeutic. These are short DNA- or RNA-based sequences that adopt a specific three-dimensional structure, enabling them to bind (and thereby inhibit) specific target molecules. One such product (Macugen) has been approved to date. RNA interference (RNAi) represents a yet additional mechanism of achieving downregulation of gene expression (Chapter 14). It shares many characteristics with antisense technology and, like antisense, provides a potential means of treating medical conditions triggered or exacerbated by the inappropriate overexpression of specific gene products. Despite the disappointing results thus far generated by nucleic-acid-based products, future technical advances will almost certainly ensure the approval of gene therapy and antisense-based products in the intermediate to longer term future.
Technological developments in areas such as genomics, proteomics and high-throughput screening are also beginning to impact significantly upon the early stages of drug development (Chapter 4). By linking changes in gene/protein expression to various disease states, for example, these technologies will identify new drug targets for such diseases. Many/most such targets will themselves be proteins, and drugs will be designed/developed specifically to interact with. They may be protein based or (more often) low molecular mass ligands.
Additional future innovations likely to impact upon pharmaceutical biotechnology include the development of alternative product production systems, alternative methods of delivery and the development of engineered cell-based therapies, particularly stem cell therapy. As mentioned previously, protein-based biotechnology products produced to date are produced in either microbial
or in animal cell lines. Work continues on the production of such products in transgenic-based production systems, specifically either transgenic plants or animals (Chapter 5).
Virtually all therapeutic proteins must enter the blood in order to promote a therapeutic effect. Such products must usually be administered parenterally. However, research continues on the development of non-parenteral routes which may prove more convenient, less costly and obtain improved patient compliance. Alternative potential delivery routes include transdermal, nasal, oral and bucal approaches, although most progress to date has been recorded with pulmonary-based delivery systems (Chapter 4). An inhaled insulin product (‘Exubera’, Chapters 4 and 11) was approved in 2006 for the treatment of type I and II diabetes.
A small number of whole-cell-based therapeutic products have also been approved to date (Chapter 14). All contain mature, fully differentiated cells extracted from a native biological source. Improved techniques now allow the harvest of embryonic and, indeed, adult stem cells, bringing the development of stem-cell-based drugs one step closer. However, the use of stem cells to replace human cells or even entire tissues/organs remains a long term goal (Chapter 14). Overall, therefore, products of pharmaceutical biotechnology play an important role in the clinic and are likely to assume an even greater relative importance in the future.
Further reading
Books
Crommelin, D. and Sindelar, R. 2002. Pharmaceutical Biotechnology, second edition. Taylor and Francis, London, UK.
Goldberg, R. 2001. Pharmaceutical Medicine, Biotechnology and European Law. Cambridge University Press. Grindley, J. and Ogden, J. 2000. Understanding Biopharmaceuticals. Manufacturing and Regulatory Issues. Interpharm Press.
Kayser, O. and Muller, RH. 2004. Pharmaceutical Biotechnology. Wiley VCH, Weinheim, Germany.
Oxender, D. and Post, L. 1999. Novel Therapeutics from Modern Biotechnology. Springer Verlag. Spada, S. and Walsh, G. 2005. Directory of Approved Biopharmaceutical Products. CRC Press, Florida, USA.
Articles
Mayhall, E., Paffett-Lugassy N., and Zon L.I. 2004. The clinical potential of stem cells. Current Opinion in Cell Biology 16, 713–720.
Reichert, J. and Paquette, C. 2003. Therapeutic recombinant proteins: trends in US approvals 1982-2002. Current Opinion in Molecular Therapy 5, 139–147.
Reichert, J. and Pavlov, A. 2004. Recombinant therapeutics – success rates, market trends and values to 2010. Nature Biotechnology 22 , 1513–1519.
Walsh, G. 2005. Biopharmaceuticals: recent approvals and likely directions. Trends in Biotechnology 23, 553–558.
Walsh, G. 2006. Biopharmaceutical benchmarks 2006. Nature Biotechnology 24, 769–776.
Weng, Z. and DeLisi, C. 2000. Protein therapeutics: promises and challenges of the 21st century. Trends in Biotechnology 20, 29–36.
2 Protein structure
2.1 Introduction
Almost all products of modern pharmaceutical biotechnology, be they on the market or likely to gain approval in the short to intermediate term, are protein based. As such, an understanding of protein structure is central to this topic. A comprehensive treatment of the subject would easily constitute a book on its own, and many such publications are available. The aim of this chapter is to provide a basic overview of the subject in order to equip the reader with a knowledge of protein science sufficient to understand relevant concepts outlined in the remaining chapters of this book. The interested reader is also referred to the ‘Further reading’ section, which lists several excellent specialist publications in the field. Much additional information may also be sourced via the web sites mentioned within the chapter.
2.2 Overview of protein structure
Proteins are macromolecules consisting of one or more polypeptides (Table 2.1). Each polypeptide consists of a chain of amino acids linked together by peptide (amide) bonds. The exact amino acid sequence is determined by the gene coding for that specific polypeptide. When synthesized, a polypeptide chain folds up, assuming a specific three-dimensional shape (i.e. a specific conformation) that is unique to it. The conformation adopted is dependent upon the polypeptide’s amino acid sequence, and this conformation is largely stabilized by multiple, weak non-covalent interactions. Any influence (e.g. certain chemicals and heat) that disrupts such weak interactions results in disruption of the polypeptide’s native conformation, a process termed denaturation. Denaturation usually results in loss of functional activity, clearly demonstrating the dependence of protein function upon protein structure. A protein’s structure currently cannot be predicted solely from its amino acid sequence. Its conformation can, however, be determined by techniques such as X-ray diffraction and nuclear magnetic resonance (NMR) spectroscopy.
Proteins are sometimes classified as ‘simple’ or ‘conjugated’. Simple proteins consist exclusively of polypeptide chain(s) with no additional chemical components present or being required for biological activity. Conjugated proteins, in addition to their polypeptide components(s),
Table 2.1 Selected examples of proteins. The number of polypeptide chains and amino acid residues constituting the protein are listed, along with its molecular mass and biological function
Protein
No. polypeptide chains
Total no. amino acids
Molecular mass (Da)Biological function
Insulin (human)2515 800Complex, includes regulation of blood glucose levels
Lysozyme (egg)112913 900Enzyme capable of degrading peptidoglycan in bacterial cell walls
IL-2 (human)113315 400T-lymphocytederived polypeptide that regulates many aspects of immunity
EPO (human)116536 000Hormone that stimulates red blood cell production
Hexokinase (yeast)2800102 000Enzyme capable of phosphorylating selected monosaccharides
Glutamate dehydrogenase (bovine) 40 8 300 1 000 000Enzyme interconverts glutamate and α -ketoglutarate and NH4+
contain one or more non-polypeptide constituents known as prosthetic group(s). The most common prosthetic groups found in association with proteins include carbohydrates (glycoproteins), phosphate groups (phosphoproteins), vitamin derivatives (e.g. flavoproteins) and metal ions (metalloproteins).
Table 2.2 The 20 commonly occurring amino acids. They may be subdivided into five groups on the basis of side-chain structure. Their three- and one-letter abbreviations are also listed (one-letter abbreviations are generally used only when compiling extended sequence data, mainly to minimize writing space and effort). In addition to their individual molecular masses, the percentage occurrence of each amino acid in an ‘average’ protein is also presented. These data were generated from sequence analysis of over 1000 different proteins
R group classificationAmino acid
Nonpolar, aliphatic
Abbreviation
Occurrence in ‘average’ protein (%) 3 letters1 letter
Molecular mass
GlycineGlyG757.2
AlanineAlaA898.3
ValineValV1176.6
LeucineLeuL1319
IsoleucineIleI1315.2
ProlineProP1155.1
AromaticTyrosineTyrY1813.2
PhenylalaninePheF1653.9
TryptophanTrpW2041.3
Polar but uncharged
Positively charged
CysteineCysC1211.7
SerineSerS1056
MethionineMetM1492.4
ThreonineThrT1195.8
AsparagineAsnN1324.4
GlutamineGlnQ1464
ArginineArgR1745.7
LysineLysK1465.7
HistidineHisH1552.2
Negatively charged Aspartic acidAspD1335.3
Glutamic acidGluE1476.2
2.2.1
Primary structure
Polypeptides are linear, unbranched polymers, potentially containing up to 20 different monomer types (i.e. the 20 commonly occurring amino acids) linked together in a precise predefined sequence. The primary structure of a polypeptide refers to its exact amino acid sequence, along with the exact positioning of any disulfide bonds present (described later). The 20 commonly occurring amino acids are listed in Table 2.2, along with their abbreviated and one-letter designations. The structures of these amino acids are presented in Figure 2.1. Nineteen of these amino acids contain a central (α) carbon atom, to which is attached a hydrogen atom (H), an amino group (NH2) a carboxyl group (COOH), and an additional side chain (R) group – which differs from amino acid to amino acid. The amino acid proline is unusual in that its R group forms a direct covalent bond with the nitrogen atom of what is the free amino group in other amino acids (Figure 2.1).
Figure 2.1 The chemical structure of the 20 amino acids commonly found in proteins
As will be evident from Section 2.2.2, peptide bond formation between adjacent amino acid residues entails the establishment of covalent linkages between the amino and carboxyl groups attached to their respective central (α) carbon atoms. Hence, the free functional (i.e. chemically reactive) groups in polypeptides are almost entirely present as part of the constituent amino acids’ side chains (R groups). In addition to determining the chemical reactivity of a polypeptide, these R groups also very largely dictate the final conformation adopted by a polypeptide. Stabilizing/repulsive forces between different R groups (as well as between R groups and the surrounding aqueous media) largely dictate what final shape the polypeptide adopts, as will be described later.
Tryptophan Tyrosine
Phenylalanine
Glycine
LeucineIsoleucine Proline
The R groups of the non-polar, alipathic amino acids (Gly, Ala, Val, Leu, Ile and Pro) are devoid of chemically reactive functional groups. These R groups are noteworthy in that, when present in a polypeptide’s backbone, they tend to interact with each other non-covalently (via hydrophobic interactions). These interactions have a significant stabilizing influence on protein conformation.
Glycine is noteworthy in that its R group is a hydrogen atom. This means that the α-carbon of glycine is not asymmetric, i.e. is not a chiral centre. (To be a chiral centre the carbon would have to have four different chemical groups attached to it; in this case, two of its four attached groups are identical.) As a consequence, glycine does not occur in multiple stereo-isomeric forms, unlike the remaining amino acids, which occur as either D or L isomers. Only L-amino acids are naturally found in polypeptides.
The side chains of the aromatic amino acids (Phe, Tyr and Trp) are not particularly reactive chemically, but they all absorb ultraviolet (UV) light. Tyr and Trp in particular absorb strongly at 280 nm, allowing detection and quantification of proteins in solution by measuring the absorbance at this wavelength.
Of the six polar but uncharged amino acids, two (cysteine and methionine) are unusual in that they contain a sulfur atom. The side chain of methionine is non-polar and relatively unreactive, although the sulfur atom is susceptible to oxidation. In contrast, the thiol ( C SH) portion of cysteine’s R group is the most reactive functional group of any amino acid side chain. In vivo, this group can form complexes with various metal ions and is readily oxidized, forming ‘disulfide linkages’ (covalent linkages between two cysteine residues within the same or even different polypeptide backbones). These help stabilize the three-dimensional structure of such polypeptides. Interchain disulfide linkages can also form, in which cysteines from two different polypeptides participate. This is a very effective way of covalently linking adjacent polypeptides.
Of the four remaining polar but uncharged amino acids, the R groups of serine and threonine contain hydroxyl (OH) groups and the R groups of asparagine and glutamine contain amide (CONH2) groups. None are particularly reactive chemically; however, upon exposure to high temperatures or extremes of pH, the latter two can deamidate, yielding aspartic acid and glutamic acid respectively.
Aspartic and glutamic acids are themselves negatively charged under physiological conditions. This allows them to chelate certain metal ions, and also to markedly influence the conformation adopted by polypeptide chains in which they are found.
Lysine, arganine and histidine are positively charged amino acids. The arganine R group consists of a hydrophobic chain of four CH2 groups ( Figure 2.1), capped with an amino (NH2) group, which is ionized (NH3 ) under most physiological conditions. However, within most polypeptides there is normally a fraction of un-ionized lysines, and these (unlike their ionized counterparts) are quite chemically reactive. Such lysine side chains can be chemically converted into various analogues. The arganine side chain is also quite bulky, consisting of three CH2 groups, an amino group ( NH2) and an ionized guanido group ( " NH2 ). The ‘imidazole’ side chain of histidine can be described chemically as a tertiary amine (R3 N), and thus it can act as a strong nucleophilic catalyst (the nitrogen atom houses a lone pair of electrons, making it a ‘nucleus lover’ or nucleophile; it can donate its electron pair to an ‘electron lover’ or electrophile). As such, the histidine side chain often constitute an essential part of some enzyme active sites.
In addition to the 20 ‘common’ amino acids, some modified amino acids are also found in several proteins. These amino acids are normally altered via a process of post-translational modification (PTM) reactions (i.e. modified after protein synthesis is complete). Almost 200 such modified amino acids have been characterized to date. The more common such modifications are discussed separately in Section 2.5.
Peptide bond
bond
Figure 2.2 (a) Peptide bond formation. (b) Polypeptides consist of a linear chain of amino acids successively linked via peptide bonds. (c) The peptide bond displays partial double-bonded character
2.2.2 The peptide bond
Successive amino acids are joined together during protein synthesis via a ‘peptide’ (i.e. amide) bond ( Figure 2.2). This is a condensation reaction, as a water molecule is eliminated during bond formation. Each amino acid in the resultant polypeptide is termed a ‘residue’, and the polypeptide chain will display a free amino (NH2) group at one end and a free carboxyl (COOH) group at the other end. These are termed the amino and carboxyl termini respectively.
The peptide bond has a rigid, planar structure and is in the region of 1.33 Å in length. Its rigid nature is a reflection of the fact that the amide nitrogen lone pair of electrons is delocalized across the bond (i.e. the bond structure is a halfway house between the two forms illustrated in Figure 2.2c). In most instances, peptide groups assume a ‘trans’ configuration ( Figure 2.2b). This minimizes steric interference between the R groups of successive amino acid residues.
Peptide
Amino acid 'residue'
Planar (rigid) peptide bonds
N - C α bond free to rotate, angle of rotation = φ
Cα - C bond free to rotate, angle of rotation = ψ
Figure 2.3 Fragment of polypeptide chain backbone illustrating rigid peptide bonds and the intervening N C α and C α C backbone linkages, which are free to rotate
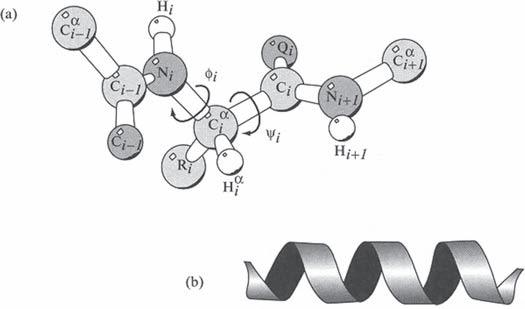
Whereas the peptide bond is rigid, the other two bond types found in the polypeptide backbone (i.e. the N Cα bond and the Cα C bond, Figure 2.3) are free to rotate. The polypeptide backbone can thus be viewed as a series of planar ‘plates’ that can rotate relative to one another. The angle of rotation around the N Cα bond is termed φ (phi) and that around the Cα C bond is termed ψ (psi) ( Figure 2.3). These angles are also known as rotation angles, dihedral angles or torsion angles. By convention, these angles are defined as being 180 when the polypeptide chain is in its fully extended, trans form. In principle, each bond can rotate to any value between 180 and 180 . However, the degrees of rotation actually observed are restricted due to the occurrence of steric hindrance between atoms of the polypeptide backbone and those of amino acid side chains. For each amino acid residue in a polypeptide backbone, the actual φ and ψ angles that are physically possible can be calculated, and these angle pairs are often plotted against each other in a diagram termed a Ramachandran plot. Sterically allowable angles fall within relatively narrow bands in most instances. A greater than average degree of φ /ψ rotational freedom is observed around glycine residues, due to the latter’s small R group – hence steric hindrance is minimized. On the other hand, bond angle freedom around proline residues is quite restricted due to this amino acid’s unusual structure (Figure 2.1). The φ and ψ angles allowable around each Cα in a polypeptide backbone obviously exert a major influence upon the final three-dimensional shape assumed by the polypeptide.
2.2.3 Amino acid sequence determination
The amino acid sequence of a polypeptide may be determined directly via chemical sequencing or by physical fragmentation and analysis, usually by mass spectrometry. Direct chemical sequencing was the only method available until the 1970s. Insulin was the first protein to be sequenced by this approach (in 1953), requiring several years and several hundred grams of protein to complete. The method has been refined and automated over the years, such that, today, polypeptides containing 100 amino acids or more can be automatically sequenced within a few hours, using microgram to milligram levels of protein. The actual chemical sequencing procedure employed is termed the Edman degradation method.
Table 2.3 Representative organisms whose genomes have been or will soon be completely/ almost completely sequenced. Data taken largely from http://wit.integratedgenomics.com/GOLD/ eucaryoticgenomes.html and http://www.tigr.org/tdb/mdb/mdcomplete.html. Updated information is available on these sites
An alternative approach to amino acid sequence determination is to sequence its gene (Chapter 3). The amino acid sequence can be inferred from the nucleotide sequence obtained. This approach has gained favour in recent years. Refinements to DNA sequencing methodologies and equipment have made such sequence analysis both rapid and relatively inexpensive. The ongoing genome projects continue to generate enormous amounts of sequence data. By the early 2000s, substantial/complete sequence data for some 300 organisms were available ( Table 2.3). As a result, the putative amino acid sequences of an enormous number of proteins (most of unknown function/structure) had been determined.
Upon its generation, sequence information is normally submitted to various databases. The major databases in which protein primary sequence data are available are listed in Table 2.4. Also included in this table are the major nucleic acid sequence databases, as amino acid sequence information can potentially be derived from these.
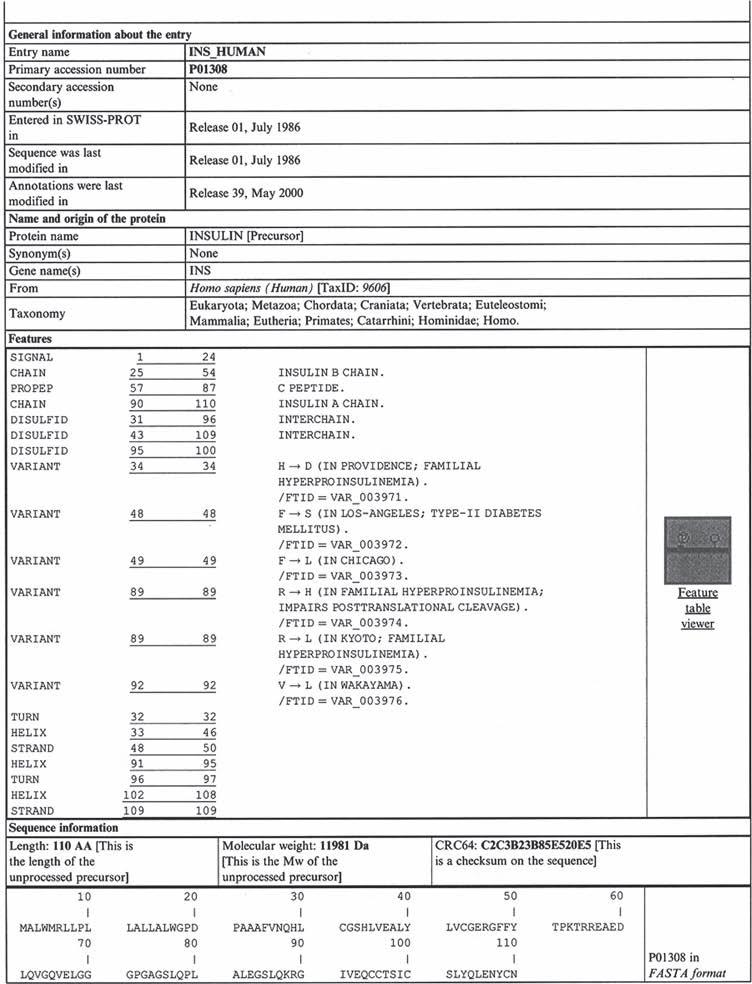
The Swiss-Prot database is probably the most widely used protein database. It is maintained collaboratively by the European Bioinformatics Institute (EBI) and the Swiss Institute for Bioinformatics. It is relatively easy to access and search via the World Wide Web (Table 2.4). A sample entry for human insulin is provided in Figure 2.4. Additional information detailing such databases is available via the web addresses provided in Table 2.4 and in the bioinformatics publications listed at the end of this chapter.
A polypeptide’s amino acid sequence can thus be determined by direct chemical (Edman) or physical (mass spectrometry) means, or indirectly via gene sequencing. In practice, these methods are complementary to one another and can be used to cross-check sequence accuracy. If the target gene/messenger RNA (mRNA) has been previously isolated, then DNA sequencing is usually most convenient. However, this approach reveals little information regarding any PTMs present in the mature polypeptide, many of whom are of critical significance in the context of therapeutic proteins (discussed in Section 2.5).
Figure 2.4 Sample entry for human insulin as present in the Swiss-Prot database. Refer to text for further details. Reproduced from the Swiss-Prot database on the Uniprot website htt://www.ebi.uniprot.org/
2.2.4 Polypeptide synthesis
Full-scale polypeptide characterization usually requires modest/large (milligram to gram) amounts of the purified target polypeptide. Even larger quantities are then generally required
if the polypeptide has a commercial application. In some cases a polypeptide can be obtained in sufficient quantities by direct extraction from its natural producer source. However, polypeptides may also be produced by direct chemical synthesis, as long as their amino acid sequence (and any PTMs) has been elucidated. Synthesis can be undertaken via a biological route (recombinant DNA technology), as is the case for virtually all modern therapeutic proteins.
2.3 Higher level structure
Thus far we have concentrated on the primary structure (amino acid sequence) of a polypeptide. Higher level protein structure can be described at various levels, i.e. secondary, tertiary and quaternary:
Secondary structure can be described as the local spatial conformation of a polypeptide’s backbone, excluding the constituent amino acid’s side chains. The major elements of secondary structure are the α -helix and β -strands, as described below.
Tertiary structure refers to the three-dimensional arrangement of all the atoms that contribute to the polypeptide.
Quaternary structure refers to the overall spatial arrangement of polypeptide subunits within a protein composed of two or more polypeptides.
2.3.1 Secondary structure
By studying the backbone of most proteins, stretches of amino acids that adopt a regular, recurring shape usually become evident. The most commonly observed secondary structural elements are termed the α -helix and β -strands, which are usually separated by stretches largely devoid of regular, recurring conformation. The α -helix and β -sheets are commonly formed because they maximize formation of stabilizing intramolecular hydrogen bonds and minimize steric repulsion between adjacent side chain groups, while also being compatible with the rigid planar nature of the peptide bonds.
The α -helix contains 3.6 amino acid residues in a full turn ( Figure 2.5). This approximates to a length of 0.56 nm along the long axis of the helix. The participating amino acid side chains protrude outward from the helical backbone. Amino acids most conducive with α -helix formation include alanine, leucine, methionine and glutamate. Proline, as well as the occurrence in close proximity of multiple residues with either bulky side groups or side groups of the same charge, tends to disrupt α -helical formation. The helical structure is stabilized by hydrogen bonding, with every backbone C = O group forming a hydrogen bond with the N—H group four residues ahead of it in the helix. Stretches of α -helix found in globular (i.e. tightly folded, approximately spherical) polypeptides can vary in length from a single helical turn to greater than 10 consecutive helical turns. The average length is about three turns.
Figure 2.5 Ball-and-stick and ribbon representations of an α -helix. Reproduced from Sun, P. and Boyington. 1997. Current Protocols in Protein Science by kind permission of the publisher, John Wiley and Sons
Stretches of α -helix are most often positioned on the protein’s surface, with one face of the helix facing the hydrophobic interior and the other facing the surrounding aqueous medium. The amino acid sequence of these helices is such that hydrophobic amino acid residues are positioned on one
Figure 2.6 The β -sheet. (a) Two segments of β -strands (antiparallel) forming a β -sheet via hydrogen bonding. The β -strand is drawn schematically as a thick arrow. By convention the arrowhead points in the direction of the polypeptide’s C terminus. (b) Schematic illustration of a two-strand β -sheet in parallel and antiparallel modes