Kreislauf- und Stoffwechselerkrankungen in Klinik und Praxis
Jahrgang 39, Heft 1
März 2026
VERL AG PERFUSION
Offizielles Organ der Deutschen Gesellschaft für Arterioskleroseforschung
Current Contents/ Clinical Medicine
ÜBERSICHTSARBEIT
Safran, ein Wundermittel?
FOREN
Forum diabeticum:
• Semaglutid-spezifische Wirkung auf kardiorenale Risiken ergänzt bekannte HbA1c- und Gewichtsreduktion
• Betazellen schützen: Früherkennung von Typ-1-Diabetes als erster wichtiger Schritt
• Hohe Zufriedenheit mit Insulin glargin 300 E/ml
Forum Lipidsenker: Modernes Lipidmanagement: Mit Alirocumab das Erreichen der Leitlinienziele fördern
Forum Elektrolytstörungen: Chronische Hyperkaliämie: Nationaler Konsensus empfiehlt moderne Kaliumbinder
Forum cardiologicum:
• SCOUT-HCM zeigt die Wirksamkeit von Mavacamten bei Jugendlichen mit HOCM
• Finerenon für Patienten mit Herzinsuffizienz mit LFEF ≥40 % zur Zulassung in der EU empfohlen
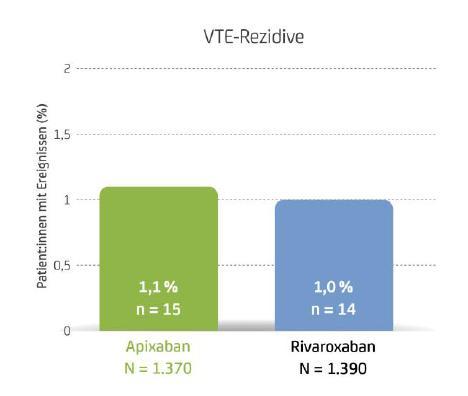
Forum antithromboticum: COBRRA – die erste Head-to-Head-Studie der NOACs Apixaban und Rivaroxaban bei akuter venöser Thromboembolie
REDAKTIONELLER TEIL
Mitteilungen, Kongressberichte
PRALUENT® ,
na klar!
NEXT GEN' PRALUENT® PEN
Aus Gründen der besseren Lesbarkeit wird auf die gleichzeitige Verwendung der männlichen, weiblichen und diversen Sprachformen verzichtet. * Die Wertschöpfung der „NextGen PRALUENT® Pens“ erfolgt in deutscher Fertigung; ‡ Es wurde auch eine nur nominal statistisch signifikante Reduktion der Gesamtmortalität bei hierarchischer Testung beobachtet (HR 0,85; 95 %-KI: 0,73–0,98). Die Ergebnisse beziehen sich auf Patienten mit stattgehabtem akutem Koronarsyndrom1;♥ Subkutane Injektion 300 mg alle vier Wochen (monatlich)1; § PRALUENT® steht in drei Dosierungen zur Verfügung. Die empfohlenen Dosen von Alirocumab betragen 75 mg einmal alle zwei Wochen, 150 mg einmal alle zwei Wochen, 300 mg einmal alle vier Wochen (monatlich), subkutan verabreicht. Alle Dosierungen können zur Einleitung der Behandlung verwendet werden;1 ¥ Der Fertigpen von PRALUENT® wurde im Vergleich zu früheren Pens in Bezug auf taktile Eigenschaften und bei einzelnen Darreichungsformen bzgl. der Anwendungsschritte optimiert. U. a. können jetzt alle PRALUENT® Dosierungen mit Pens vergleichbarer Bauart injiziert werden – dem PRALUENT® Pen der nächsten Generation („NextGen PRALUENT® Pen“). HR = Hazard Ratio; KI = Konfidenzintervall; LDL-C = Lipoprotein-Cholesterin niederer Dichte; NEXT GEN ' = Next Generation. 1 Fachinformation Praluent®, Stand 10/2025. Praluent® 75/150/300 mg Injektionslösung im Fertigpen Wirkstoff: Alirocumab. Zusammensetzung: Arzneilich wirksame Bestandteile: Ein Fertigpen zur einmaligen Anwendung enthält 75/150 mg Alirocumab in 1 ml Lösung; ein Fertigpen zur einmaligen Anwendung enthält 300 mg Alirocumab in 2 ml Lösung. Alirocumab ist ein humaner monoklonaler IgG1-Antikörper, der mittels rekombinanter DNA-Technologie aus Ovarialzellen des chinesischen Hamsters (CHO-Zellen) gewonnen wird. Sonstige Bestandteile: Histidin, Saccharose, Polysorbat 20, H2O für Injektionszwecke. Anwendungsgebiete: Primäre Hypercholesterinämie und gemischte Dyslipidämie: Begleitend zu einer Diät bei Erwachsenen mit primärer Hypercholesterinämie oder gemischter Dyslipidämie und bei Kindern und Jugendlichen im Alter von 8 Jahren und älter mit heterozygoter familiärer Hypercholesterinämie – in Kombination mit einem Statin oder mit einem Statin und anderen lipidsenkenden Therapien bei Patienten, die mit einer maximal verträglichen Statin-Therapie die LDL-C-Zielwerte nicht erreichen, oder – als Monotherapie oder in Kombination mit anderen lipidsenkenden Therapien bei Patienten mit einer Statin-Unverträglichkeit oder wenn Statine kontraindiziert sind. Bestehende atherosklerotische kardiovaskuläre Erkrankung: Bei Erwachsenen mit bestehender atherosklerotischer kardiovaskulärer Erkrankung zur Reduktion des kardiovaskulären Risikos durch Verringerung der LDL-C-Werte zusätzlich zur Korrektur anderer Risikofaktoren – in Kombination mit einer maximal verträglichen Statin-Therapie mit oder ohne anderen lipidsenkenden Therapieprinzipien oder – als Monotherapie oder in Kombination mit anderen lipidsenkenden Therapieprinzipien bei Patienten mit einer Statin-Unverträglichkeit oder wenn Statine kontraindiziert sind. Zu Studienergebnissen bezüglich der Wirksamkeit auf LDL-C, kardiovaskuläre Ereignisse und die untersuchten Populationen siehe Abschnitt 5.1. der Fachinformation. Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile. Nebenwirkungen: Immunsystem: Selten: Überempfindlichkeit, Hypersensibilitätsvaskulitis. Atemwege/Brust/ Mediastinum: Häufig: klinische Zeichen und Symptome im Bereich der oberen Atemwege. Haut/Unterhautgewebe: Häufig: Pruritus; selten: Urtikaria, nummuläres Ekzem; nicht bekannt: Angioödem. Allgemeine Beschwerden am Verabreichungsort: Häufig: Reaktionen an der Injektionsstelle; nicht bekannt: grippeartige Erkrankung. Abgabe/Verschreibungspflicht: Deutschland: Verschreibungspflichtig. Österreich: Rezept- und apothekenpflichtig, wiederholte Abgabe verboten. Pharmakotherapeutische Gruppe/ATC-Code: Mittel, die den Lipidstoffwechsel beeinflussen. Andere Mittel, die den Lipidstoffwechsel beeinflussen. ATC-Code: C10AX14. Pharmazeutischer Unternehmer: Sanofi Winthrop Industrie, 82 avenue Raspail, 94250 Gentilly, Frankreich. Örtlicher Vertreter des Zulassungsinhabers: Sanofi-Aventis Deutschland GmbH, 65926 Frankfurt am Main, Deutschland. sanofi-aventis GmbH, 1100 Wien, Österreich.
Stand der Information: Oktober 2025
Weitere Angaben zu den besonderen Warnhinweisen und Vorsichtsmaßnahmen für die Anwendung, Wechselwirkungen mit anderen Arzneimitteln und sonstige Wechselwirkungen, Fertilität, Schwangerschaft und Stillzeit, Nebenwirkungen sowie ggf. Gewöhnungseffekten sind der veröffentlichten Fachinformation zu entnehmen.
Sanofi-Aventis Deutschland GmbH, Lützowstr. 107, 10785 Berlin
Entmenschlichte Medizin
Ich habe soeben ein Buch publiziert, von dem ich meine, es sei wichtig. Daher erlaube ich mir, die Einleitung in leicht modifizierter Form hier wiederzugeben, sodass Sie sich ein Bild vom Inhalt machen können:
Im Jahr 1990 wurde ich auf einen Lehrstuhl der Medizinischen Fakultät der Universität Wien berufen. Gleichzeitig erhielt ich, ein gebürtiger Deutscher, die österreichische Staatsbürgerschaft, denn damals war es nur Österreichern erlaubt, in Wien Ordinarius zu werden. Ich nahm an, dass ich für den Rest meiner Laufbahn in Wien bleiben würde. Dass es dann doch anders kam, liegt nicht zuletzt an dem Thema dieses Buches.
Meine erste und wichtigste Aufgabe in Wien war es, eine Gruppe von rund 20 Mitarbeitern auf etwa 100 zu vergrößern und gleichzeitig mein rasch wachsendes Team als erste klinische Abteilung vom altehrwürdigen Allgemeinen Krankenhaus (AKH) in ein brandneues Gebäude zu übersiedeln. Als es schließlich so weit war und wir das neue AKH in Betrieb nehmen konnten, wurde dieser Tag für Österreich ein durchaus historisches Ereignis. Neben den vielen Ansprachen der Politiker vor laufenden Kameras sollte auch ich als Klinikchef etwas Passendes zum Besten geben.
Man hatte mir ausreichend Vorwarnung gegeben, sodass ich Zeit und Muße hatte, ein gutes Thema für diese Gelegenheit zu finden. Spontan dachte ich zunächst daran, die Geschichte meiner Klinik und des AKH zu beleuchten und somit den Tag in einen historischen Kontext zu stellen. Wie sich bald herausstellte, war das keine gute Idee. Die recherchierbare Vergangenheit riss mit dem Jahr 1938 abrupt ab. Diesbezügliche Dokumente waren aus den Bibliotheken verschwunden, und wenn ich jemanden um Auskunft bat, wurde mir mit Stirnrunzeln zu verstehen gegeben, dass man an diesem Thema wohl besser nicht rühre. Welches Thema? Natürlich, wie konnte ich es nur vergessen: Der „Anschluss“ von 1938! Ich entschloss mich daher, zur Eröff-
nung des AKH lediglich ein paar unverfängliche Worte zu sprechen und meine Recherchen zur NS-Vergangenheit meiner Klinik und der Medizinischen Fakultät zu vertagen. Die diesbezüglichen Nachforschungen haben mir dann meinen Verbleib in Wien zunehmend schwer gemacht. Im Jahr 1993, nahm ich daher einen Ruf ins englische Exeter an, und 1995 publizierte ich meinen Bericht unter dem Titel „A leading medical school seriously damaged: Vienna 1938“ [1]. Obschon es in Exeter meine Aufgabe wurde, die sogenannte Alternativmedizin zu erforschen, hat mich das „Nazi-Thema“ seither nicht mehr losgelassen. Wann immer meine Zeit es erlaubte, forschte ich in diesem Bereich. Was mich dabei besonders faszinierte und bedrückte, war die Einsicht, dass mein Berufsstand nicht etwa nur ein neutraler Mitläufer gewesen war, sondern sich bereitwilligst und aktiv an den Untaten des Dritten Reichs beteiligt hatte.
Um es ganz klar auszudrücken: Ohne die Mediziner wäre ein Großteil der Gräueltaten des Dritten Reichs nicht möglich gewesen. Ärzte hatten das Konzept der Rassenhygiene entwickelt, es begeistert aufgegriffen und sodann auch gnadenlos umgesetzt. Der Abstieg in die Unmenschlichkeit begann mit der Zwangssterilisation derjenigen, die von den Nazis unter rassenhygienischen Gesichtspunkten als genetisch minderwertig eingestuft worden waren. Es folgte die Pervertierung des Konzepts der Euthanasie zu einem Programm der Tötung behinderter Patienten. Schließlich wurde der Massenmord an Juden und KZHäftlingen, die wegen Erschöpfung als
unnützer Ballast galten, geplant und exekutiert. Den Gipfel der Barbarei stellten letztlich unfassbar grausame Menschenversuche von SS-Medizinern dar, die sich als Wissenschaftler verstanden, jedoch nichts weiter als unmenschliche Berserker waren. Diese Ansammlung ärztlicher Untaten stellt mit Abstand den schwersten Verstoß gegen die Ethik und Moral in der Geschichte der Medizin dar. Erich Kästner hat es auf den Punkt gebracht, als er zum Dritten Reich kommentierte: „Ein besonders widerwärtiges Kapitel in der Geschichtsschreibung … wird den Ärzten gewidmet werden müssen.“ Während diese Medizin-Verbrechen, wenn auch nur zögerlich, in Deutschland langsam aufgearbeitet wurden, schien man in Österreich in den 1990er Jahren noch eine andere Strategie zu bevorzugen. Hier herrschte zu diesem Thema meist ein eisiges Schweigen. Falls eine Diskussion dennoch unvermeidbar war, so versuchte man in Österreich meist in die Opferrolle zu schlüpfen. Man berief sich nur allzu gerne auf die Moskauer Deklaration von 1943, die besagte, dass Österreich das erste Opfer der Angriffspolitik Hitlers war. Eine offene und systematische Auseinandersetzung mit der NS-Vergangenheit wurde so gut es ging verhindert. Forscher, die hier nicht mitspielen wollten, wurden als Nestbeschmutzer gebrandmarkt. Seit meinem Weggang aus Wien hat sich diese Situation in erfreulicher Weise gebessert, und es gibt zahlreiche Ansätze einer rühmlichen Aufarbeitung der unrühmlichen Rolle der österreichischen Ärzteschaft im Dritten Reich. Ein Kompendium mit biographischen Skizzen derjenigen Mediziner, die sich auf die eine oder andere Weise an den NS-Verbrechen beteiligt haben, existiert jedoch bis heute nicht. Mein Buch stellt den Versuch dar, diese Lücke zu schließen.
Edzard Ernst, Emeritus Professor, University of Exeter
Quellen:
1 Ernst E. A leading medical school seriously damaged: Vienna 1938. Ann Intern Med 1995;122:789-792
2 Kaestner E: Notabene 45. Fischer 1965
Prof. Dr. med. E. Ernst, Exeter, U.K.
Offizielles Organ
Deutschen Gesellschaft für Arterioskleroseforschung
Medicine
Ozempic ® 2 mg. Stärker denn je.1
Stärkere
HbA1c - und Gewichtssenkung1
Starke
Evidenz 1,3
Stark fürs Herz 2
Jetzt Ozempic® verordnen für noch mehr Stärke1
PZN 20298705
Mehr zu Ozempic® ? QR-Code scannen! www.novo-wissen.de/ ozempic-2mg
Ozempic® ist wirtschaftlich mit einem AMNOG verhandelten, transparenten Preis
Zum Basistext: QR-Code scannen. novo-wissen.de/bt-ozempic
1. Frías JP et al. Lancet Diabetes Endocrinol 2021;9:563–574. Einzelheiten zur Studie: SUSTAIN FORTE war eine 40-wöchige, randomisierte, doppelblinde, multizentrische, multinationale, parallelgruppen Phase-3b Studie, in der die Wirksamkeit und Sicherheit von einmal wöchentlich verabreichtem Semaglutid 2,0 mg gegenüber Semaglutid 1,0 mg bei erwachsenen Patienten mit Typ 2 Diabetes untersucht wurde. Patienten wurden 1:1 randomisiert auf Semaglutid 2,0 mg bzw. Semaglutid 1,0 mg. Eingeschlossen wurden erwachsene Patienten mit Typ 2 Diabetes, die eine unzureichende glykämische Kontrolle hatten (HbA1c-Einschlussbereich in der Studie) und mit stabiler antidiabetischer Therapie (inkl. Metformin erlaubt) behandelt wurden; der mittlere Ausgangs-HbA1c lag im Studienkollektiv bei 8,9 %. Ziel der Studie war, die Überlegenheit von Semaglutid 2,0 mg gegenüber Semaglutid 1,0 mg bezüglich HbA1c-Senkung nach 40 Wochen zu prüfen sowie Sicherheits- und Verträglichkeitsprofile zu vergleichen. Der primäre Endpunkt war die Veränderung des HbA1c gegenüber Baseline nach 40 Wochen. Zu den vordefinierten sekundären Endpunkten zählten die Veränderung des Körpergewichts gegenüber Baseline, der Anteil der Patienten, die bestimmte HbA1c-Ziele (z. B. < 7,0 %) erreichten, sowie Sicherheits- und Verträglichkeitsparameter (einschließlich gastrointestinaler Nebenwirkungen und schwerwiegender unerwünschter Ereignisse). Bei Semaglutid 2,0 mg zeigte sich für den HbA1c-Wert eine mittlere Senkung von – 2,2 % und eine mittlere Gewichtsreduktion von –6,9 kg. 2. Tan X et al. Diabetes Obes Metab 2026;1–11. 3. Rodbard HW et al. Efficacy and safety of once-weekly semaglutide 2.0 mg as an add-on to dose-reduced insulin glargine vs titrated insulin glargine in people with type 2 diabetes and overweight: SUSTAIN OPTIMIZE. EASD 61st Annual Meeting. September 18th 2025. Einzelheiten zur Studie: SUSTAIN OPTIMIZE war eine 40-wöchige, randomisierte, offene, multizentrische, multinationale, parallelgruppen Phase-3b Studie mit erwachsenen Patienten, die einen unzureichend kontrollierten Typ 2 Diabetes (HbA1c 7–10 %) und einen BMI von ≥ 25 kg/m2 aufwiesen sowie einmal täglich Basalinsulin (≤ 40 E/Tag) und Metformin, mit oder ohne SGLT-2 Inhibitoren, erhielten. Patienten wurden auf eine reduzierte Insulin Glargin E 100 Dosis einmal täglich und 2 mg Semaglutid einmal wöchentlich oder Insulin Glargin E 100 (titriert) einmal täglich randomisiert. Primärer Endpunkt war die Veränderung des HbA1c-Werts vom Ausgangswert bis Woche 40. Im sekundären Endpunkt wurde u.a. die Veränderung des Körpergewichts vom Ausgangswert bis Woche 40 untersucht. Hier konnte eine Gewichtssenkung bis zu –8,0 kg gezeigt werden. Ozempic® ist eine eingetragene Marke der Novo Nordisk A/S, Dänemark.
E. Ernst: Safran, ein Wundermittel?
ÜBERSICHTSARBEIT
Safran, ein Wundermittel?
Edzard Ernst Emeritus Professor, University of Exeter, UK
PERFUSION 2026; 01: 4–7
Safran (Crocus sativus) wird seit Jahrhunderten als Heilmittel angepriesen. Heute wird er sogar oft als Allheilmittel deklariert. Aber natürlich sind derartige Annahmen kein Ersatz für wissenschaftliche Evidenz. Die Pflanze enthält Crocetin, ein Aglykon von Crocin, das natürlich in Safran vorkommt. Crocetin wird eine breite Palette von Wirkungen zugeschrieben, z.B. kardioprotektive, hepatoprotektive, neuroprotektive, antidepressive, antivirale, krebshemmende, antiatherosklerotische, antidiabetische und gedächtnisfördernde Eigenschaften. Die wichtigere Frage ist jedoch: Wie gut ist die klinische Evidenz?
Zu Safran existiert heute eine erstaunliche Anzahl klinischer Studien. Da Einzelstudien uns jedoch leicht in die Irre führen können, ist es vielleicht besser, wenn wir uns auf gut gemachte Zusammenfassungen solcher Untersuchungen verlassen. Im Folgenden sind die Ergebnisse einiger aktuellen systematischen Übersichtsarbeiten zu den wichtigsten Anwendungsgebieten aufgeführt:
Depression und Angstsymptome
Die Meta-Analyse von 8 Studien zur Beurteilung depressiver Symptome
Zusammenfassung
Safran (Crocus sativus) wird als traditionelles Heilmittel genutzt und heute als Allheilmittel angepriesen. In dieser Übersicht werden aktuelle systematische Übersichten zu unterschiedlichen Indikationen zusammengefasst, aus denen sich insgesamt ein eher konsistent positives Bild ergibt. Gleichzeitig wird hervorgehoben, dass die Primärstudien oft klein, kurz, heterogen und methodisch limitiert sind, und auch die Qualität der Reviews variiert. Insgesamt erscheint Safran daher als interessantes pflanzliches Mittel mit breitem, aber noch nicht abschließend gesichertem therapeutischem Potenzial, das insbesondere größere, hochwertige Studien verdient.
Saffron (Crocus sativus) has long been promoted as a medicinal plant and is nowadays portrayed as a panacea. Recent years have seen a surprising number of randomized trials, and several systematic reviews and meta-analyses now summarize its effects across a broad range of indications. However, many primary studies are often small, short-term and methodologically heterogeneous, and the quality of the reviews is variable, which limits the strength of the conclusions. Overall, saffron emerges as a promising herbal intervention with consistently positive but still preliminary clinical evidence, warranting larger, high-quality trials before firm therapeutic recommendations can be made.
ergab keinen signifikanten Unterschied zwischen Safran und SSRIs bei der Reduktion depressiver Symptome. Vier Studien zu Angstsym-
ptomen zeigten ebenfalls keinen signifikanten Unterschied zwischen Safran und SSRIs bei der Reduktion von Angstsymptomen. Hinsichtlich
E. Ernst: Safran, ein Wundermittel?
der Sicherheit beklagten die Studienteilnehmer der Safran-Gruppe weniger unerwünschte Ereignisse als die in der SSRI-Gruppe. Die Autoren schlussfolgerten, dass Safran eine potenzielle Alternative zu SSRIs sei, um depressive und Angstsymptome mit weniger unerwünschten Ereignissen zu reduzieren [1].
Prämenstruelles Syndrom (PMS) und Dysmenorrhö
Safran zeigte einen signifikant positiven Effekt auf PMS-Symptome. Darüber hinaus war Safran auch wirksam bei der Reduktion der Dysmenorrhö. Die Autoren zogen den Schluss, dass Safran günstige Wirkungen auf die Symptome von PMS und Dysmenorrhö bei Frauen ausübt [2].
Vier Studien erfüllten die Einschlusskriterien dieses Reviews mit insgesamt 118 Patienten. Die Ergebnisse deuten auf eine effiziente Rolle von Safran als adjuvante Therapie oder als Monotherapie bei ADHS hin. Ernste Nebenwirkungen wurden nicht beobachtet. Die Autoren schlussfolgerten, dass Safran vielversprechend bei der Verbesserung von ADHS-Symptomen ist und ein akzeptables Sicherheitsprofil aufweist [3].
Neurologische und psychiatrische Erkrankungen
In diese Zusammenfassung wurden 46 randomisierte Studien eingeschlossen. Die Behandlungsdauer
lag bei 4 – 48 Wochen. Safran erwies sich als effektiver als Placebo bezüglich der Verbesserung von Kognition, Depression, Angst und Schlafstörungen. Die Schlussfolgerung der Autoren: Safran war den konventionellen Medikamenten nicht unterlegen bei der Behandlung kognitiver Störungen, Depression, Angst, ADHS und Zwangsstörungen und zeigte eine gute Verträglichkeit mit wenigen Nebenwirkungen [4].
Diabetes mellitus
Zehn randomisierte Studien mit insgesamt 562 Teilnehmern wurden in diese Meta-Analyse eingeschlossen. Safran wurde in Dosen von 5 mg/Tag bis 1 g/Tag verabreicht. Im Vergleich zu Placebo reduzierte die Safran-Supplementierung signifikant den Nüchternblutzucker und den HbA1c-Wert, zeigte jedoch keinen signifikanten Effekt auf den Insulinspiegel. Safran erwies sich somit bei Diabetes-Patienten als wirksam und stellt ein vielversprechendes adjuvantes Mittel zur glykämischen Kontrolle dar. Aufgrund der heterogenen methodischen Qualität der Studien bleibt die Interpretation des Nutzens jedoch eingeschränkt. Daher sind längere Follow-ups sowie groß angelegte, qualitativ hochwertige klinische Studien erforderlich, um definitive Empfehlungen zu geben [5].
Störungen des weiblichen Fortpflanzungssystems
Insgesamt wurden 50 Studien zum Einfluss von Safran auf das weibliche Fortpflanzungssystem identifiziert. Diese belegen die Wirksamkeit
von Safran oder seinen Inhaltsstoffen bei der Regulation von Sexualhormonen, der Förderung der Follikelentwicklung und Ovulation sowie dem Schutz von Eierstöcken und Uterus vor oxidativem Stress. Zudem lindern sie Symptome von Dysmenorrhö, prämenstruellem Syndrom, Menopause, polyzystischem Ovarialsyndrom (PCOS) und sexueller Dysfunktion. Safran gilt ferner als vielversprechender Kandidat für Studien oder klinische Prüfungen zu Ovarial- und Zervixkarzinomen. Die Schlussfolgerung der Autoren lautete: Crocus sativus verbessert die Symptome diverser Störungen des weiblichen Fortpflanzungssystems, was vor allem auf sekundäre Pflanzenstoffe wie Crocin, Crocetin und Safranal zurückzuführen ist [6].
Schlafstörungen
Acht Studien mit insgesamt 431 Teilnehmern wurden in diese Analyse eingeschlossen. Im Vergleich zu Placebo reduzierte Safran die Schwere der Schlaflosigkeit und verbesserte die Schlafqualität sowie die Schlafdauer signifikant. Die Autoren zogen den Schluss, dass Safran trotz begrenzter Evidenzqualität den an Insomnie leidenden Patienten helfen kann. Diese Option reduziert potenziell den Bedarf an sedierend-hypnotischen Medikamenten und damit das Risiko von Abhängigkeit sowie Entzugserscheinungen [7].
Nierenfunktion
Neun randomisierte Studien wurden in die Meta-Analyse eingeschlos-
sen. Ihre Qualität wurde mit dem Cochrane-Risiko-Bias-Tool bewertet. Die gepoolte Analyse zeigte, dass eine Safran-Supplementierung im Vergleich zu Placebo keinen signifikanten Effekt auf die Serumharnstoffkonzentration und den Serumkreatininspiegel ausübt. In der Dosis-Wirkungs-Analyse zeigte sich eine signifikante nichtlineare Beziehung zwischen der Dauer der Safran-Supplementierung und den Serumharnstoff- und Kreatininspiegeln. Somit hat eine SafranSupplementierung keine negativen Auswirkungen auf Nierenfunktionsmarker [8].
Kardiovaskuläre Risikofaktoren
Für diese Analyse wurden 32 Studien mit einer Gesamtfallzahl von 1674 berücksichtigt. Safran reduzierte signifikant die Triglyzeride (TG), das Gesamtcholesterin (TC), LDL, den Nüchternblutzuckerspiegel (FBG), den HbA1c-Wert, den HOMA-IR-Wert (Homeostasis Model Assessment), den systolischen Blutdruck (SBP), TNF-α, Taillenumfang (WC), Malondialdehyd (MDA)und Alanintransferase (ALT). Außerdem erhöhte Safran die totale Antioxidantienkapazität (TAC). Es zeigte sich eine lineare Regression zwischen FBG und der Dauer der Safranaufnahme. Die nichtlineare Dosis-Wirkungs-Analyse zeigte signifikante Assoziationen mit HDL, HOMA-IR, Gewicht, ALP, FBG, HbA1c und TNF-α. Eine nichtlineare Assoziation ergab sich zwischen der Interventionsdauer und HDL sowie DBP. Die Autoren schlussfolgerten, dass Safran TG, TC, LDL, FBG, HbA1c, HOMAIR, SBP, CRP, TNF-α, WC, MDA,
TAC und ALT effektiv verbessern kann [9].
Leberfunktion
Acht Studien mit insgesamt 463 Teilnehmern wurden in diese systematische Übersichtsarbeit eingeschlossen. Safran war mit einem statistisch signifikanten Rückgang der Aspartataminotransferase (AST) im Vergleich zu Placebo assoziiert. Safran hatte keinen signifikanten Effekt auf die Alaninaminotransferase (ALT)- und Alkalische Phosphatase (ALP)-Spiegel. Safran zeigte somit günstige Auswirkungen auf die Leberfunktion [10].
Blutdruck
Acht randomisierte Studien wurden in diese Zusammenfassung eingeschlossen. Die Safran-Supplementierung führte zu einem signifikanten Rückgang des systolischen und des diastolischen Blutdrucks (DBP). Safran reduzierte den DBP nichtlinear in Abhängigkeit von der Dauer. Eine Safran-Supplementierung kann daher den systolischen und diastolischen Blutdruck bei Erwachsenen signifikant verbessern. Es sollte jedoch beachtet werden, dass die blutdrucksenkenden Effekte der Safran-Supplementierung gering waren und möglicherweise nur eine geringe klinische Bedeutung haben [11].
Körpergewicht und Lipide
Insgesamt wurden 14 Studien in diese Meta-Analyse aufgenommen. Es fand sich eine signifikan-
te Reduktion von Cholesterin und Triglyzeriden (TG) nach Safran-Intervention, aber kein signifikanter Effekt auf Gewicht und LDL-Konzentration. Eine Meta-Regressionsanalyse zeigte, dass eine langfristige Safran-Supplementierung die HDL-Spiegel erhöhen kann. Die Ergebnisse deuten somit auf einige Vorteile von Safran hinsichtlich Cholesterin, HDL und TG im Vergleich zu Placebo hin [12].
Sexualstörungen bei Männern und Frauen
Insgesamt 5 Studien mit 173 Teilnehmern wurden in diese MetaAnalyse eingeschlossen. Die Ergebnisse zeigten einen statistisch signifikanten positiven Effekt von Safran auf sexuelle Funktionsstörungen. Daher schlossen die Autoren, dass Safran die sexuelle Dysfunktion wirksam verbessern kann [13].
Verhaltensstörungen
Zwölf Studien erfüllten die Einschlusskriterien dieser Analyse. Sie untersuchten die Effekte von Safran auf psychologische/verhaltensbezogene Outcomes bei schwerer depressiver Störung (n = 6), prämenstruellem Syndrom (n = 1), sexueller Dysfunktion und Infertilität (n = 4) sowie Gewichtsverlust/ Snackverhalten (n = 1). Die Ergebnisse zeigen die Wirksamkeit von Safran im Vergleich zu Placebo bei der Verbesserung depressiver Symptome (im Vergleich zu Antidepressiva und Placebo), prämenstrueller Symptome und sexueller Dysfunktion. Zusätzlich war Safran auch
E. Ernst: Safran, ein Wundermittel?
E. Ernst: Safran, ein Wundermittel?
wirksam bei der Reduktion exzessiven Snackverhaltens [14].
Fazit
Zweifellos lassen sich an der methodischen Qualität der Primärstudien erhebliche Defizite bemängeln, und auch die zahlreichen Reviews – von denen hier nur eine Auswahl vorgestellt wurde – entsprechen nicht durchgängig dem höchsten Standard. Dennoch verleihen diese Übersichtsarbeiten dem Safran – nicht zuletzt durch ihre weitgehend einheitlich positiven Bewertungen – das Profil eines durchaus interessanten pflanzlichen Mittels, das weitere, sorgfältig geplante Forschung verdient. Hinzu kommen abschließend zwei weitere Aspekte: Safran zeigt bei sachgemäßer Dosierung praktisch keine ernsthaften Nebenwirkungen, sodass das Nutzen-Risiko-Profil insgesamt als günstig einzustufen ist. Zudem sind die Kosten für Safran-Präparate eher niedrig, da hierfür andere Pflanzenteile verwendet werden können statt der erheblich wertvolleren Griffelnarben, aus denen das klassische Safran-Gewürz gewonnen wird.
Literatur
1 Shafiee A et al. Effect of saffron versus selective serotonin reuptake inhibitors (SSRIs) in treatment of depression and anxiety: a meta-analysis of randomized controlled trials. Nutr Rev 2025; 83:e751-e761
2 Mohammadi MM et al. Effect of saffron on premenstrual syndrome and dysmenorrhea: a systematic review and meta-analysis. Korean J Fam Med 2026;47:69-80
3 Seyedi-Sahebari S et al. The effects of Crocus sativus (Saffron) on ADHD: a systematic review. J Atten Disord 2024; 28:14-24
4 Han S et al. New horizons for the study of saffron (Crocus sativus L.) and its active ingredients in the management of neurological and psychiatric disorders: a systematic review of clinical evidence and mechanisms. Phytother Res 2024; 38:2276-2302
5 Liu J et al. Effect of saffron supplementation on the glycemic outcomes in diabetes: a systematic review and metaanalysis. Front Nutr 2024;11:1349006
6 Hasheminasab FS et al. Therapeutic effects of saffron (Crocus sativus L) on female reproductive system disorders: a systematic review. Phytother Res 2024; 38:2832-2846
7 Munirah MP et al. Crocus sativus for insomnia: a systematic review and meta-analysis. Int J Environ Res Public Health 2022;19:11658
8 Norouzy A et al. The effects of saffron supplementation on the measures of renal function indicators: a systematic review and meta-analysis. Int Urol Nephrol 2022;54:2215-2226
9 Zamani M et al. The effects of saffron supplementation on cardiovascular risk factors in adults: a systematic review and dose-response meta-analysis. Front Nutr 2022;9:1055517
10 Hasani M et al. Effect of saffron supplementation on liver enzymes: a systematic review and meta-analysis of randomized controlled trials. Diabetes Metab Syndr 2021;15:102311
11 Setayesh L et al. The effect of saffron supplementation on blood pressure in adults: a systematic review and dose-response meta-analysis of randomized controlled trials. Nutrients. 2021;13: 2736
12 Rahmani J et al. The effect of saffron on weight and lipid profile: a systematic review, meta-analysis, and dose-response of randomized clinical trials. Phytother Res 2019;33:2244-2255
13 Ranjbar H et al. Effects of saffron (Crocus sativus) on sexual dysfunction among men and women: A systematic review and meta-analysis. Avicenna J Phytomed 2019;9:419-427
14 Hausenblas HA et al. A systematic review of randomized controlled trials examining the effectiveness of saffron (Crocus sativus L.) on psychological and behavioral outcomes. J Integr Med 2015;13:231-240
Anschrift des Verfassers: Prof. Edzard Ernst, MD, PhD, FMEdSci, FRSB, FRCP, FRCP(Edin.) Emeritus Professor, University of Exeter, UK E.Ernst@exeter.ac.uk
MITTEILUNGEN
Neu zur Behandlung von PID, SID und CIDP: Hizentra® 50-ml-Fertigspritze mit 10 g IgG
Hizentra® ist derzeit das einzige in Deutschland verfügbare subkutane Immunglobulin (SCIg) in Fertigspritzenform. Die 10 g/50 ml-Option ergänzt das bestehende Portfolio (2 g/10 ml, 4 g/20 ml) um ein „Großformat“ für Patienten, die bei chronisch inflammatorisch demyelinisierender Polyneuropathie (CIDP) oder bei primären und sekundären Immundefekten (PID/ SID) eine hohe Dosierung benötigen. Fertigspritzen können die Vorbereitungszeit verkürzen, die Handhabung erleichtern und sind mit einer hohen Therapiezufriedenheit assoziiert.
Indiziert ist die neue 10 g/50 mlFertigspritze:
• bei: PID wie der X-chromosomalen Agammaglobulinämie (XLA) oder dem variablen Immundefektsyndrom (CVID), die eine hochdosierte Substitution erfordern, oder bei höherem Körpergewicht, höheren angestrebten IgG-Zielspiegeln oder verlängerten Dosierungsintervallen,
• bei CIDP aufgrund von immunmodulatorischen Erhaltungsdosen, die sich in der Praxis häufig in größere Wochenmengen übersetzen.
Einfache Anwendung im Alltag
Nach der Therapieeinleitung und Schulung durch qualifiziertes medizinisches Fachpersonal können Patienten die Fertigspritzen in der Heimselbsttherapie anwenden. Die 50-ml-Fertigspritze eignet sich für die pumpenassistierte subkutane Infusion. Eine einfache Anwendung wird vor allem durch zwei Faktoren ermöglicht: Eine kontrollierte Silikonierung trägt dazu bei, dass die Gleitkräfte des Kolbens über die Zeit konstant bleiben – ein relevanter Faktor für die Handhabung. Zum anderen lässt sich die IgG-Lösung mithilfe des Tip-toTip-Connectors in die pumpengeeignete Spritze überführen – ein Vorteil insbesondere für Menschen mit eingeschränkter Handmotorik. Eine summative Human-FactorStudie* untersuchte die Anwendung der Hizentra® 50-ml-Fertigspritze mit Infusionspumpe unter praxisnahen Bedingungen und zeigte: 99 % der Teilnehmer (n = 80/81) konnten die Infusionsdosis vollständig und erfolgreich verabreichen. Bei den kritischen Anwendungsschritten lag die Erfolgsrate bei mindestens 90 %. Viele Fehler bezogen sich auf das Zubehör und nicht auf die Gebrauchstauglichkeit der Fertigspritze selbst.
Weniger Zeitaufwand, weniger Schritte, mehr Therapiezufriedenheit
Eine aktuelle Befragung** von kanadischen Patienten mit PID und SID verglich die SCIg-Verabreichung mit Fertigspritzen (5 ml und 10 ml) und Durchstechflaschen (Vials). Im Ergebnis konnten Fertigspritzen die Heimselbstinfusion und den Therapiealltag spürbar erleichtern. Patienten berichteten über kürzere Vorbereitungszeiten, weniger Behandlungsschritte und eine tendenziell kürzere Infusionsdauer – Faktoren, die die Therapiezufriedenheit und die Adhärenz in der Routine mitprägen.
Fazit für die Praxis
Die neue 10 g/50 ml-Fertigspritze kann insbesondere bei höheren Dosierungen die Zahl der benötigten Einheiten pro Gabe reduzieren, Vorbereitungsschritte weiter straffen und damit die Anwendung bei pumpenassistierter Infusion zusätzlich erleichtern.
S. M.
* Dhruv et al. Präsentation auf der 13. Jahrestagung der Immunoglobulin National Society in Washington D.C., 2024
** Mallick R et al. BMC Immunol 2024; 25:18
Helga und Rudolf gehören auf den Schlitten.
Nicht ins
Krankenhaus.
Mit FORXIGA® das Hospitalisierungsrisiko bei HF signifikant reduzieren.1,a
HF = chronische Herzinsuffizienz.
a Das relative Risiko war in dem präspezifizierten Endpunkt Gesamtzahl der Hospitalisierungen aufgrund von Herzinsuffizienz in der gepoolten Meta-Analyse der Primärdaten von DAPA-HF2 und DELIVER3 signifikant reduziert ohne Hinweis auf Effektmodifikation der einzelnen Ejektionsfraktionen.1 1 Vgl. Jhund PS et al. Nat Med 2022; 28(9):1956–1964. 2 Vgl. McMurray JJV et al. N Engl J Med 2019; 381(21):1995–2008 (inkl. Supplement). 3 Vgl. Solomon SD et al. N Engl J Med 2022; 387(12):1089–1098 (inkl. Supplement).
Forxiga® 5 mg Filmtabletten, Forxiga® 10 mg Filmtabletten. Wirkstoff: Dapagliflozin. Verschreibungspflichtig. Zusammensetzung: 1 Filmtablette Forxiga® 5 mg enthält Dapagliflozin-(2S)-Propan-1,2-diol (1:1) (1 H2O), entsprechend 5 mg Dapagliflozin. 1 Filmtablette Forxiga® 10 mg enthält Dapagliflozin-(2S)Propan-1,2-diol (1:1) (1 H2O), entsprechend 10 mg Dapagliflozin. Sonstige Bestandteile: Tablettenkern: Mikrokristalline Cellulose, Lactose, Crospovidon, Siliciumdioxid, Magnesiumstearat. Filmüberzug: Poly(vinylalkohol), Titandioxid, Macrogol 3350, Talkum, Eisen(III)-hydroxid-oxid x H2O. Anwendungsgebiete: Indiziert bei Erwachsenen und Kindern im Alter von 10 Jahren und älter zur Behandlung von unzureichend kontrolliertem Typ-2-Diabetes mellitus in Ergänzung zu einer Diät und Bewegung als Monotherapie, wenn Metformin aufgrund einer Unverträglichkeit als ungeeignet erachtet wird; zusätzlich zu anderen Arzneimitteln zur Behandlung des Typ-2-Diabetes. Zu Studienergebnissen im Hinblick auf Kombinationen von Behandlungen, die Wirkung auf die Blutzuckerkontrolle, kardiovaskuläre und renale Ereignisse sowie die untersuchten Populationen, siehe Abschnitte 4.4, 4.5 und 5.1 der Fachinformation. Patienten mit schwerer Leberfunktionsstörung: Es wird eine Anfangsdosis von 5 mg empfohlen. Wenn diese gut vertragen wird, kann die Dosis auf 10 mg erhöht werden. Herzinsuffizienz: Indiziert bei erwachsenen Patienten zur Behandlung der symptomatischen, chronischen Herzinsuffizienz. Chronische Niereninsuffizienz: Indiziert bei erwachsenen Patienten zur Behandlung der chronischen Niereninsuffizienz. Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile. Nebenwirkungen: Sehr häufig: Hypoglykämie (bei Anwendung mit einem Sulfonylharnstoff oder Insulin). Häufig: Vulvovaginitis, Balanitis und verwandte Infektionen des Genitalbereichs, Harnwegsinfektionen, Schwindel, Hautausschlag, Rückenschmerzen, Dysurie, Polyurie, erhöhter Hämatokrit, verminderte renale Kreatinin-Clearance zu Behandlungsbeginn, Dyslipidämie. Gelegentlich: Pilzinfektionen, Volumenmangel, Durst, Verstopfung, Mundtrockenheit, Nykturie, vulvovaginaler Pruritus, Pruritus genitalis, erhöhtes Kreatinin im Blut zu Behandlungsbeginn, erhöhter Harnstoff im Blut, Gewichtsreduktion. Selten: diabetische Ketoazidose (bei Typ-2-Diabetes). Sehr selten: nekrotisierende Fasziitis des Perineums (Fournier-Gangrän), Angioödem, tubulointerstitielle Nephritis. Weitere Hinweise: siehe Fachinformationen. Pharmazeutischer Unternehmer: AstraZeneca GmbH, Friesenweg 26, 22763 Hamburg, E-Mail: azinfo@astrazeneca.com, www.astrazeneca.de, Servicehotline für Produktanfragen: 0800 22 88 660. Stand: August 2024.
Der Diabetes Typ 2 bleibt ein zentraler Risikofaktor für Herz-Kreislauf- und Nierenerkrankungen. Eine Behandlung mit GLP-1 Rezeptoragonisten (GLP-1 RA) kann jedoch über die Blutzucker- und Gewichtssenkung hinaus dazu beitragen, das Risiko für diese Begleiterkrankungen wirksam zu senken [1]. Auf der DDG-Herbsttagung 2025 wurden aktuelle Studiendaten zum GLP-1 RA Semaglutid (Ozempic®, 1 wöchentlich [2]) vorgestellt, die über die bekannten positiven Effekte hinsichtlich der Blutzucker- und Gewichtsreduktion hinausgehen. Sie zeigen in Klinik [3, 4] und Praxis [5] eine effektive Risikoreduktion für kardiovaskuläre [3, 5] und renale [4] Ereignisse sowie funktionelle Verbesserungen bei pAVK [6].
Klinisch relevante Blutzuckerund Gewichtsreduktion –evidenzbasiert überlegen und in der Praxis erreichbar
Daten aus einer direkten Vergleichsstudie mit Semaglutid und Dulaglutid zeigen einen überlegenen Effekt von Semaglutid 1 mg auf HbA1c und Gewicht: –1,8 % HbA1c-Reduktion versus –1,4 % unter Dula-
FORUM DIABETICUM
Semaglutid-spezifische Wirkung auf kardiorenale Risiken ergänzt bekannte HbA1c- und Gewichtsreduktion
glutid 1,5 mg (Estimated Treatment Difference [ETD]: –0,41; 95%-KI: –0,57 bis –0,25; p < 0,0001) sowie –6,5 kg Gewichtsreduktion versus –3 kg unter Dulaglutid 1,5 mg (ETD: –3,55; 95%-KI: –4,32 bis –2,78; p < 0,0001) [7]*.
Kardioprotektive Wirkung von Semaglutid:
Zusatznutzen in CV-Endpunktstudie wird durch Real-WorldDaten ergänzt
Ozempic® ist das einzige Inkretinmimetikum mit einem vom Gemeinsamen Bundesausschuss (G-BA) anerkannten kardiovaskulären Zusatznutzen [8]. Grundlage hierfür waren die Ergebnisse der kardiovaskulären Endpunktstudie SUSTAIN 6, in der Semaglutid 1 mg eine 26%ige relative Risikoreduktion für schwere kardiovaskuläre Ereignisse (MACE) gegenüber Placebo zeigte [3]. Diese Evidenz wird nun durch aktuelle RealWorld-Daten aus der REACH-Studie ergänzt [5].
* Senkung des HbA1c-Werts und des Gewichts, berechnet ausgehend von Baseline, verglichen gegenüber Woche 40 für Semaglutid 1 mg und Dulaglutid 1,5 mg.
In der REACH-Analyse überzeugte der GLP-1 RA Semaglutid im Vergleich zu Dulaglutid durch eine überlegene Wirksamkeit bei Patienten mit Typ-2-Diabetes und atherosklerotischer Herz-Kreislauf-Erkrankung (ASCVD): Die mit Semaglutid behandelten Studienteilnehmer hatten ein um 23 % geringeres Risiko für Herzinfarkt, Schlaganfall oder Tod jeglicher Ursache (3-Punkte-MACE) im Vergleich zu Dulaglutid [5]. Dabei war das Risiko für Schlaganfälle um 20 % reduziert [5].
Nachgewiesene Vorteile von Semaglutid bei Typ-2-Diabetes, CKD und pAVK
In FLOW, der weltweit ersten Nierenendpunkt-Studie mit einem GLP-1 RA, wurde bei Patienten mit Typ-2-Diabetes und vorbestehender chronischer Nierenerkrankung (CKD) das Risiko für schwere Nierenereignisse unter Semaglutid 1 mg versus Placebo um 24 % reduziert (HR: 0,76; 95%-KI: 0,66 – 0,88; p = 0,0003) [4]. Ebenso sank die mittlere jährliche Abnahme der geschätzten glomerulären Filtrationsrate (eGFR) um durchschnittlich 1,16 ml/min/1,73m2/
FORUM DIABETICUM
Semaglutid
Semaglutid (Ozempic®) ist ein 1 x pro Woche injizierbarer GLP-1 Rezeptoragonist, zu dessen Wirksamkeit und Sicherheit Erfahrungen aus über 21 Millionen Patientenjahren vorliegen [10]. Ozempic® ist zugelassen zur Behandlung des unzureichend kontrollierten Diabetes mellitus Typ 2 bei Erwachsenen als Zusatz zu Diät und körperlicher Aktivität als Monotherapie, wenn die Anwendung von Metformin aufgrund einer Unverträglichkeit oder Kontraindikation ungeeignet ist, oder zusätzlich zu anderen Arzneimitteln zur Behandlung des Diabetes mellitus [2].
Jahr (95%-KI: 0,86 – 1,47; p < 0,001) [4]. Basierend auf diesen Ergebnissen erhielt Ozempic® als bisher einziges Inkretinmimetikum eine Leitlinienempfehlung bei Typ2-Diabetes und CKD [9]. Daten der STRIDE-Studie zeigen außerdem, dass die Behandlung mit Semaglutid 1 mg die Funktionalität und Lebensqualität von Patienten mit Typ-2-Diabetes und pAVK signifikant verbessern kann. Die Studie erreichte ihren primären Endpunkt: Unter Semaglutid konnte im Vergleich zu Placebo nach 52 Wochen die maximale Gehstrecke bei einer Steigung von 12 % um 13 % erhöht werden [6]. Dies entspricht einer durchschnittlichen Verlängerung um 39,9 Meter und wird als klinisch bedeutsame Verbesserung eingestuft. Darüber hinaus zeigte sich eine Überlegenheit von Semaglutid gegenüber Placebo auch bei der schmerzfreien Gehstrecke, bei der gesundheitsbezogenen Lebensqualität, gemessen anhand des
Vascular Quality of Life Questionnaire-6 (VascuQoL-6) sowie bei der maximalen Gehstrecke nach 57 Wochen [6].
Semaglutid als Therapie mit breitem kardiometabolischem Nutzen
Die Daten aus SUSTAIN 6 [3] und REACH [5] sowie FLOW [4] und STRIDE [6] untermauern die zentrale Rolle von Semaglutid in der modernen Behandlung des Typ2-Diabetes. Der GLP-1 RA trägt nicht nur zur Reduktion des kardiovaskulären [3, 5] und renalen [4] Risikos bei, sondern verbessert auch die körperliche Leistungsfähigkeit und Lebensqualität von Patienten mit Begleiterkrankungen wie der pAVK [6] – und damit deren Alltag und Prognose gleichermaßen.
Elisabeth Wilhelmi, München
Literatur
1 Bundesärztekammer (BÄK), Kassenärztliche Bundesvereinigung (KBV), Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF). Nationale Versorgungsleitlinie Typ-2-Diabetes – Version 3.0. 2023 AWMF-Register-Nr. nvl-001, https://register.awmf.org/de/leitlinien/detail/nvl-001
2 Fachinformation Ozempic®, aktueller Stand.
3 Marso SP et al. N Engl J Med 2016; 375:1834-1844
4 Perkovic V et al. N Engl J Med 2024; 391:109-121 (+Suppl.)
7 Pratley RE et al. Lancet Diabetes Endocrinol 2018;6:275-286
8 Anhaltspunkt für einen geringen Zusatznutzen bei erwachsenen Patienten mit T2D und manifester CV Erkrankung, siehe Beschlusstext G-BA Anlage XII der AM-RL vom 2. Mai 2019 Absatz 1) b.) 2b.). Bestätigt am 20.03.2025
9 Diabetes Care 2025;48(Suppl 1):S181S206
10 Interne Berechnung Novo Nordisk IQVIA Ozempic® Patientenjahre in Deutschland seit Markteinführung einschließlich Februar 2025
Früher erkennen, gezielter behandeln, langfristig schützen – darum geht es beim Thema Früherkennung von Typ-1-Diabetes (T1D). Dr. Franziska Schaaff, Fachärztin für Kinder- und Jugendmedizin, Gemeinschaftspraxis für Kinderund Jugendmedizin Eckental, gab im Rahmen einer von Sanofi veranstalteten Fachpressekonferenz Tipps zur Integration der T1DFrüherkennung in den Praxisalltag und erläuterte die klinische Relevanz der Betazell-Restfunktion zum Zeitpunkt der Diagnose. Anschließend fasste Dr. Christiane Look, Leiterin der medizinischen Abteilung General Medicines GSA bei Sanofi, wichtige Meilensteine im Zulassungsprozess von Teplizumab zusammen.
Eine chronische Autoimmunerkrankung mit Langzeitfolgen
Die Ursachen des Typ-1-Diabetes sind bis heute nicht alle bekannt. Die Betazell-Schädigung setzt bereits Jahre vor der Hyperglykämie ein [1]. Aktuell unterscheidet man 4 Stadien der Autoimmunerkrankung: Stadium 1 und 2 verlaufen meist unbemerkt. Erst ab Stadium 3 führt der starke Betazellverlust zu klinischen Symptomen und einer messbaren Hyperglykämie. „Eine exogene Insulingabe ist nun unverzichtbar“, erläuterte Schaaff [2]. Stadium 4 kennzeichnet den Langzeit-T1D [3].
Die Patienten erkranken oft schon in jungen Jahren. In Deutschland ist etwa eines von 425 Kindern
FORUM DIABETICUM
Betazellen schützen:
Früherkennung von Typ-1-Diabetes als erster wichtiger Schritt
und Jugendlichen von T1D betroffen [4]. Betroffene Kinder müssen den Großteil ihres Lebens mit der chronischen Erkrankung leben. Dies geht u.a. mit erheblichen psychosozialen Begleiterscheinungen durch Ängste und alltägliche Einschränkungen einher, so die Ärztin.
Ohne Vorwarnung ist die Erstmanifestation zudem häufig von Komplikationen wie einer diabetischen Ketoazidose (DKA) begleitet [5]. Die DKA-Rate ist in den letzten Jahren gestiegen – besonders häufig sind Kinder betroffen. Bis zu 40 % davon manifestieren mit einer ausgeprägten Stoffwechselentgleisung [6]. „T1D ist eine gefährliche Erkrankung im Kindesalter. Das müssen wir uns immer wieder bewusst machen“, betonte Schaaff. Auch für die behandelnden Ärzte ist eine DKA eine Stresssituation. „In dieser Situation kann viel schief gehen, was mit schwerwiegenden Langzeitfolgen verbunden ist. Eine DKA sollte unbedingt vermieden werden“, mahnte sie.
Jede Betazelle zählt
Trotz deutlicher Fortschritte im Krankheitsmanagement mit Technologien wie dem kontinuierlichen Glukosemonitoring (CGM) und
den Insulinpumpen (AID) kann die Insulinersatztherapie die Feinregulation durch die Betazellen nicht ersetzen [1, 7]. Denn diese spielen nicht nur eine wichtige Rolle bei der endogenen Insulinsekretion, sondern auch bei der Regulierung der Blutzuckerwerte im Zusammenspiel mit anderen Pankreaszellen [8, 9, 10]. Dem Erhalt der Betazellen kommt daher eine hohe Bedeutung zu.
Als Maß für die Betazellfunktion kann das C-Peptid dienen, das in den Betazellen im gleichen Verhältnis mit Insulin freigesetzt wird und somit direkt auf die Restfunktion schließen lässt [11]. Studien zeigen, dass eine höhere BetazellRestfunktion mit einem geringeren Insulinbedarf [12], niedrigeren HbA1c-Werten [13], einer niedrigeren Zahl schwerer Hypoglykämien [14] und einem geringeren Risiko für Langzeitfolgen wie Retinopathie und Nephropathie assoziiert ist [15].
„Die moderne Insulintherapie ist kein Ersatz für die komplexe Homöostase des Pankreas. Wenn Betazellen erhalten werden, wird auch die natürliche Regulation zwischen Insulin und Glukagon erhalten. Hierdurch können Hyper- und Hypoglykämien besser vermieden werden“, machte Schaaff deutlich.
Früherkennung in der Kinderarztpraxis –praxistauglich und wirksam
Die Früherkennung spielt eine entscheidende Rolle bei der Prävention und frühen Intervention – und sie beginnt in der Kinderarztpraxis. Laut Schaaff kann ein T1D bereits Monate bis Jahre vor der klinischen Manifestation über Inselautoantikörper im Blut nachgewiesen werden. Liegen mindestens 2 Autoantikörper vor, spricht man von einem T1D-Frühstadium [1]. In Deutschland können Kinder zwischen 2 und 10 Jahren im Rahmen der vom Helmholtz Munich initiierten Fr1da-Studie getestet werden [16]. Momentan wird Fr1da in folgenden Bundesländern angeboten: Bayern, Rheinland-Pfalz, Hessen, Niedersachsen, Hamburg und Sachsen. „Die Ausweitung soll nun beschleunigt werden. Bis Ende 2026 ist eine Abdeckung von über 80 % anvisiert und wir bei Sanofi unterstützen diese Initiative natürlich“, berichtete Look. Ergänzend bietet „Fr1da für Verwandte“ eine deutschlandweite Früherkennungsuntersuchung auf Autoantikörper für Angehörige ersten und zweiten Grades (bis 21 Jahre) [17]. Diese haben ein deutlich erhöhtes Risiko, selbst an T1D zu erkranken [18]. „Unsere Praxis hat von Beginn an bei Fr1da teilgenommen“, berichtete Dr. Schaaff. „Der Informationszettel zur Studie liegt jeder Vorsorgemappe bis zur U11 bei. Bei uns wird die Früherkennung sowohl bei den Vorsorgeuntersuchungen als auch in normalen Sprechstundenterminen angeboten.“ Wichtig
FORUM DIABETICUM
Mehr Informationen zum Thema Früherkennung des Typ-1-Diabetes finden Sie unter: gemeinsam-typ1.de.
für eine erfolgreiche Umsetzung sind feste Strukturen und Zuständigkeiten bei Aufklärung, Testung, Dokumentation und Befundmitteilung. Bei einem positiven Test erfolge eine Anbindung an Ambulanzen, wo die Familien umfassend über das Krankheitsbild und Warnzeichen aufgeklärt und in der Stoffwechselüberwachung geschult werden. Auch eine psychosoziale Begleitung ist gewährleistet. „Nach mittlerweile 10 Jahren Praxiserfahrung stehe ich der Früherkennung uneingeschränkt positiv gegenüber. Man tut den Familien und Kindern etwas Gutes. Diese Chance sollten wir nutzen!“ appellierte Schaaff. Die Evidenz zu positiven Effekten der Früherkennung auf die Diabeteskontrolle ist ermutigend: Kinder mit bekanntem Frühstadium weisen bei Manifestation bessere klinische Parameter auf (niedriger HbA1c und Nüchternblutzucker, höheres CPeptid, weniger Gewichtsverlust) und die DKA-Rate sank deutlich [19]. „Der Zugewinn ist klar: bessere Kontrolle und weniger Komplikationen“, fasste Schaaff zusammen und ergänzte: „Die Möglichkeiten der frühzeitigen Blutzuckereinstellung und Überwachung wirken sich positiv auf die Gesundheit der Kinder aus und ermöglichen den Kindern einen besseren Umgang mit der Erkrankung.“
Paradigmenwechsel in der T1D-Therapie mit Teplizumab
Den Progress zu Stadium 3 und damit den Betazellverlust verzögern zu können, rückt mit der Empfehlung der Europäischen Arzneimittel Agentur (EMA) zur Zulassung von Teplizumab (Teizeild) bei Erwachsenen, Jugendlichen und Kindern ab 8 Jahren mit T1D im Stadium 2 in greifbare Nähe [20]. „Hierdurch wird ein Paradigmenwechsel im Bereich T1D eingeleitet“, berichtete Look. Teplizumab soll den Übergang in Stadium 3 verzögern, wenn bereits ein signifikanter Anteil der Betazellen zerstört ist und Symptome wie ständiger Durst, häufiges Wasserlassen, unerklärlicher Gewichtsverlust und generelle Erschöpfung auftreten. Eine Zwischenanalyse der PETITE-Studie zeigte außerdem, dass auch bei Kindern unter 8 Jahren das Sicherheitsprofil von Teplizumab konsistent mit bereits berichteten Daten ist [21]. „Diese Ergebnisse stimmen uns zuversichtlich. Gerade in dieser jungen Population, bei der die Erkrankung schneller fortschreitet, wäre es von besonderer Bedeutung, in Zukunft ebenfalls eine krankheitsverzögernde Therapieoption zur Hand zu haben“, schloss Look.
Brigitte Söllner, Erlangen
14
Literatur
1 DDG 2023. S3-Leitlinie: Diagnostik, Therapie und Verlaufskontrolle des Diabetes mellitus im Kindes- und Jugendalter, AWMF-Registernummer 057016
2 Insel RA et al. Diabetes Care 2015; 38:1964-1974
3 Haller MJ et al. Horm Res Paediatr 2024;97:529-545
4 Buchmann M et al. J Health Monit 2023;8:59-81
5 Baechle C et al. Diabetes Res Clin Pract 2023;197:110559
17 Fr1da. Teilnahme Fr1da-Studie für Verwandte deutschlandweit. https://www. typ1diabetesfrueherkennung.de/teilnahme-fr1da-studie/teilnahme-fr1dastudie-fuer-verwandtedeutschlandweit. html
18 Besser REJ et al. Pediatr Diabetes 2022;23:1175-1187
19 Hummel S et al. Diabetologia 2023; 66:1633-1642
20 European Medicines Agency. Teizeild. https://www.ema.europa.eu/en/medicines/human/EPAR/teizeild
21 Gitelman SE et al. Diabetologia 2025; doi: 10.1007/s00125-025-06586-1. Epub ahead of print
FORUM DIABETICUM
Hohe Zufriedenheit mit
Insulin
glargin 300 E/ml
Menschen mit Typ-1-Diabetes (T1D) benötigen eine effektive, sichere und zuverlässige Insulintherapie. Das Gleiche gilt für Patienten, deren Typ-2-Diabetes (T2D) mit oralen Antidiabetika und/oder GLP-1-Rezeptoragonisten nicht ausreichend kontrolliert werden kann [1].
Als Basalinsulin-Analogon der zweiten Generation besitzt Insulin glargin 300 E/ml (Toujeo®) ein flaches Wirkprofil mit langanhaltender, gleichmäßiger Blutzuckersenkung bis zu 36 Stunden [2, 3]. Es hat sich in interventionellen und nichtinterventionellen Studien unter Alltagsbedingungen bei T1D und T2D bewährt und ist mit einer hohen Patientenzufriedenheit assoziiert [4, 5, 6].
Von großem Vorteil ist, dass Patienten mit unzureichend kontrolliertem Typ-1-Diabetes unter engmaschiger Stoffwechselüberwachung von anderen Basalinsulinen auf Insulin glargin 300 E/ml umgestellt werden können [7]. Dabei benötigen Patienten, die zuvor mehrmals täglich ein Basalinsulin injizieren mussten, nur einmal pro Tag Insulin glargin 300 E/ml [3, 4]. Dies verbessert die Therapiezufriedenheit und Therapiepersistenz, wie Studien bei Menschen mit Typ-1- oder Typ-2-Diabetes belegen, die auf
Insulin glargin 300 E/ml ein- oder umgestellt wurden [4, 5, 6].
Mehr Zeit im Zielbereich nach Wechsel von Insulin detemir auf Insulin glargin 300 E/ml
Die prospektive, multizentrische Studie COMET-T [7] schloss 94 Patienten aus Deutschland, Österreich und der Schweiz ein, deren T1D mit ihrem bisherigen Basalinsulin nicht gut eingestellt war – sie hatten HbA1c-Werte von 7,5 – 10 % (im Durchschnitt 8,1 %). In 2 Subgruppen zeigten sich signifikante Vorteile für Insulin glargin 300 E/ ml. So profitierten die Studienteilnehmer mit Adipositas (BMI ≥30 kg/m²; n = 22) besonders von der Umstellung: Bei ihnen stieg die Zeit im Zielbereich (Time in Range, TIR; Zielbereich: 3,9 – 10,0 mmol/l bzw. 70 – 180 mg/dl) um 8,4 %. Und bei den Teilnehmern, die zuvor mit Insulin detemir behandelt worden waren (n = 25), erhöhte sich die TIR unter Insulin glargin 300 E/ml sogar um 10,5 % – eine relevante Verbesserung. Auch im Gesamtkollektiv hatte sich die TIR 24 Wochen nach Umstellung auf Insulin glargin 300 E/ml numerisch um 3,0 Prozentpunkte verbessert (p = 0,078; nicht signifikant) [7].
Einmal täglich Insulin glargin
300 E/ml ist genug
Für die Substitution von Insulin detemir, das seit Beginn des Jahres nicht mehr verfügbar ist, bietet sich Insulin glargin 300 E/ml als Alternative an. Patienten, die zuvor einmal täglich Insulin detemir injiziert hatten, erhalten in der Regel die gleiche Tagesdosis Insulin glargin 300 E/ml, die Umstellung erfolgt im Verhältnis 1:1. Bei zweimal täglicher Injektion von Insulin detemir hingegen werden von der bisherigen Tagesdosis 20 % abgezogen, um die neue Tagesdosis zu ermitteln. Eine engmaschige Stoffwechselüberwachung wird empfohlen. Insulin glargin 300 E/ml wird in beiden Fällen nur einmal täglich appliziert und weist als langwirksames Basalinsulin-Analogon der zweiten Generation ein flaches Wirkprofil auf [2, 3].
Die Umstellung des Basalinsulins erscheint für den Patienten zunächst nicht einfach. Für die für den Therapieerfolg nötige Adhärenz ist das Vertrauen in die neue Therapie sehr wichtig. Dabei ist von Vorteil, dass Insulin glargin 300 E/ml sehr gleichmäßig wirkt. Denn je gleichmäßiger und harmonischer ein Insulin wirkt, desto stabiler ist die Blutzuckereinstellung und desto höher ist die Akzeptanz des Patienten.
FORUM DIABETICUM
Verbesserte Therapiezufriedenheit – unabhängig vom Diabetestyp
Die Therapiezufriedenheit der Patienten unter Insulin glargin 300 E/ml wurde unter anderem in den Studien OPTIMIZE [4] bei T1D und ATOS [5] bei T2D erfragt. Als Messinstrument wurde in beiden Studien der DTSQs-Score (Diabetes Treatment Satisfaction Questionnaire, status version, 0–36 Punkte) verwendet. Damit werden neben der Zufriedenheit per se auch die Flexibilität, Verständlichkeit und Alltagstauglichkeit der eigenen Behandlung, die Wahrscheinlichkeit der weiteren Anwendung und Weiterempfehlung sowie die vom Patienten selbst wahrgenommene Hyper- und Hypoglykämien erfasst. Die prospektive Interventionsstudie OPTIMIZE schloss 94 Patienten mit T1D ein. Ihr DTSQs-Score stieg innerhalb von 24 Wochen von 24,07 auf 29,38 Punkte (ITTPopulation). Dies entspricht einer Verbesserung der Therapiezufriedenheit um relative 22 % nach Umstellung auf Insulin glargin 300 E/ ml [4].
Eine Post-hoc-Analyse der prospektiven Real-World-Beobachtungsstudie ATOS schloss die Daten von 3.656 Patienten mit T2D ein, die zuvor insulinnaiv waren und auf Insulin glargin 300 E/ml eingestellt wurden. Auch hier zeig-
te sich eine deutliche Verbesserung: 86,8 % aller Patienten berichteten nach einem Jahr über eine höhere Therapiezufriedenheit. Eine klinisch bedeutsame Verbesserung mit einem Anstieg des DTSQs-Wertes um ≥4 Punkte wurde nach 12 Monaten bei 76,0 % der Teilnehmer verzeichnet [5].
Dass die Patienten die Behandlung mit Insulin glargin 300 E/ml länger fortführen, zeigt eine weitere große Real-World-Studie. Sie untersuchte retrospektiv die Wahrscheinlichkeit eines Behandlungsabbruchs nach 12 Monaten bei der Behandlung mit Insulin glargin 300 E/ml gegenüber Insulin glargin 100 E/ml und Insulin detemir und ergab eine signifikant niedrigere Abbruchrate der Behandlung mit Insulin glargin 300 E/ml [6].
Brigitte Söllner, Erlangen
Literatur
1 Nationale Versorgungsleitlinie Typ2-Diabetes; https://register.awmf.org/ de/leitlinien/detail/nvl-001
2 Fachinformation Toujeo®; Stand: November 2023
3 Bolli GB et al. Diabetes Obes Metab 2015;17:386-394
4 Mathieu C et al. Diabetes Ther 2020; 11:495-507
5 Snoek F et al. Diabetes Obes Metab 2025;27:4011-4016
6 oussel R et al. Diabetes Ther 2020;11: 1861-1872
7 Gölz S et al. Diabetes Ther 2025;16: 121-134
Dyslipidämie-Leitlinien wie die von ESC und EAS betonen seit Jahren die präventive Bedeutung individueller LDL-C-Zielwerte, die sich am kardiovaskulären Risiko orientieren [1]. Auch das im Sommer 2025 vorgelegte Focused Update der ESC/EAS-Leitlinie zur Diagnostik und Therapie der Dyslipidämien führt dies weiter und empfiehlt eine frühe und anhaltende Senkung des Low-DensityLipoprotein-Cholesterins (LDL-C) zur Zielwerterreichung. Außerdem wurde beim LDL-C-Ziel sowie der Einstufung in die Kategorien etwas nachgeschärft und eine Gruppe mit extrem hohem Risiko ergänzt [2]. Das Grundkonzept lautet dabei nach wie vor: Je höher das Risiko, desto niedriger der LDL-C-Zielwert.
Leitlinien-Update stärkt die frühe und intensive Therapie
Für Patienten, die innerhalb von 2 Jahren ein erneutes kardiovaskuläres Ereignis erleiden, empfiehlt das Focused Update einen LDL-CZielwert <40 mg/dl (<1,0 mmol/l) bei gleichzeitiger Senkung des Ausgangswerts um ≥50 % [2]. Dabei unterstreicht das Update die frühe und intensive LDL-C-Senkung. So wird bei akutem Koronar-
FORUM LIPIDSENKER
Modernes Lipidmanagement: Mit Alirocumab das Erreichen der Leitlinienziele fördern
syndrom (ACS) erstmals für lipidsenkend therapienaive Patienten eine initiale Kombinationstherapie noch während des stationären Aufenthalts empfohlen [2]. Außerdem gibt die Leitlinie eine IA-Empfehlung für den Einsatz von PCSK9Hemmern wie z.B. Alirocumab (Praluent®) in den Fällen, in denen der individuelle LDL-C-Zielwert mit einer Kombination aus Statin und Ezetimib nicht erreicht wird [1]. Dieser beträgt für Patienten mit sehr hohem Risiko, beispielsweise für die meisten Menschen mit Diabetes und/oder bekannter atherosklerotischer kardiovaskulärer Erkrankung (ASCVD), <55 mg/dl (<1,4 mmol/l) bei einer gleichzeitigen Senkung des Ausgangswertes um ≥50 % [1, 2]. Bei Patienten in niedergelassenen Praxen liegt der LDL-C-Wert im Mittel bei 130 – 140 mg/dl [3]. Selbst wenn sich dieser Wert mit einem hochintensiven Statin um 50 % absenken lässt, wird der Zielwert von <55 mg/dl nicht erreicht. Daher sollte insbesondere bei bei Menschen mit hohem und sehr hohem kardiovaskulärem Risiko, bei denen kardiovaskuläre Ereignisse so vollständig wie möglich und so effektiv wie möglich verhindert werden müssen, frühzeitig eine intensive, kombinierte LDL-C-Senkung eingeleitet werden.
Mit Alirocumab die Leitlinienziele erreichen ...
Das ODYSSEY-Studienprogramm zeigte, dass die Behandlung mit Alirocumab den LDL-C-Wert konsequent senken und so dazu beitragen kann, die Leitlinienziele zu erreichen [4–8]. Laut aktuellen Daten gelingt das auch vor dem ersten kardiovaskulären Ereignis [6, 7]. Auf dem ESC-Kongress 2025 in Madrid wurde eine Subgruppenanalyse der ODYSSEY CHOICE-I-Studie präsentiert, in der die Monatsdosis von 300 mg Alirocumab randomisiert untersucht wurde. Die Analyse fokussierte auf ASCVD-Patienten ohne ACS. Bei ihnen betrug die LDL-C-Senkung, ähnlich wie in der Gesamtkohorte, 60 – 65 % und war über den gesamten Studienzeitraum von 48 Wochen konstant [7]. Dabei profitierten die Patienten auch von der Anwenderfreundlichkeit von Alirocumab: Der PCSK9Hemmer muss nur einmal im Monat gespritzt werden [4].
... und das kardiovaskuläre Risiko anhaltend senken
Durch den konsequenten LDL-Csenkenden Effekt von Alirocumab profitieren Patienten subgruppenübergreifend von einer Reduktion
ihres kardiovaskulären Risikos. Gezeigt hat das beispielsweise eine Post-hoc-Analyse der 5-Jahres-Daten der ODYSSEY OUTCOMESStudie, in der das relative Risiko für schwere kardiovaskuläre Ereignisse (MACE) bei Patienten mit ACS unter der Therapie mit Alirocumab um 17 % reduziert werden konnte [8]. Da Patienten mit Komorbiditäten wie Diabetes mellitus ein höheres MACE-Risiko haben [10], war der absolute Nutzen der LDLC-Senkung in dieser Subpopulation besonders hoch [9].
Eine Real-World-Studie auf Basis der US-Versichertendatenbank Optum zeigte das jetzt auch für Patienten ohne ACS in der Anamnese. Die Ergebnisse sprechen dafür, dass auch in dieser Population
FORUM LIPIDSENKER
Menschen mit Diabetes in Bezug auf das MACE-Risiko besonders stark profitierten. Konkret reduzierte Alirocumab das absolute MACERisiko über 4 Jahre in der Teilpopulation ohne Diabetes signifikant um 6,5 % (p < 0,0001), in der Teilpopulation mit Diabetes waren es sogar 9,9 % (p < 0,0001) [11].
Brigitte Söllner, Erlangen
Highlights 2025“ am 28.11.2025 in Berlin
4 Fachinformation Praluent®; Stand: Oktober 2025
5 Schwartz GG et al N Engl J Med 2018;379:2097-2107
6 Filardi PP et al. Atherosclerosis 2024; 395(Suppl 1):118408
7 Tokgözoğlu L et al. Efficacy and safety of Alirocumab once-monthly dosing in patients with atherosclerotic cardiovascular disease and without acute coronary syndrome or stroke; an ODYSSEY CHOICE I analysis. Poster presentation, ESC 2025
8 Goodman SG et al. J Am Heart Ass 2023;12:e029216
9 Ray KK et al. Lancet Diabetes Endocrinol 2019;7:618-628
Literatur
1 Mach F et al. Eur Heart J 2020;41:111188
2 Mach F et al. Eur Heart J 2025;46: 4359-4378
3 Brands JM. Präsentation auf der Fachpressekonferenz „Sanofi – Unsere
10 Marx N et al. Eur Heart J 2023;44: 4043-4140
11 Cabezas MC et al. Alirocumab is associated with fewer ischemic events in patients with or without diabetes and atherosclerotic cardiovascular disease and no prior events. EASD Annual Meeting 2025; short oral presentation 1121
BANGLADESCH: Unser Team versorgt die Wunde des 4-jährigen Rohingya, Sho . Sein Vater Anas M. beruhigt ihn.
Der jüngst von einer Gruppe führender Fachärzte der Nephrologie, Kardiologie und Inneren Medizin/ Allgemeinmedizin aus Deutschland erarbeitete Nationale Konsensus [1] bündelt die aktuelle Evidenz zum Hyperkaliämie-Management und leitet hieraus praxisnahe Empfehlungen für den Versorgungsalltag in der Kardiologie, Nephrologie und Inneren Medizin ab – ein wichtiger Schritt angesichts der bislang unzureichenden Versorgung, inklusive der Nutzung moderner Kaliumbinder [2, 3].
Anlässlich eines von AstzraZeneca ausgerichteten Pressegespräch am 02. März 2026 erläuterten die an der Publikation beteiligten Experten Dr. Birgit Schleß, PD Dr. Sven Waßmann und Prof. Prof. Dr. Markus van der Giet die Relevanz des Hyperkaliämie-Managements für den Versorgungsalltag in der Kardiologie, Nephrologie und Inneren Medizin.
Die Hyperkaliämie ist eine potenziell lebensbedrohliche Elektrolytentgleisung [4], die besonders häufig bei kardiorenalen Patienten [5, 6, 7] oder unter der – in diesem Kontext gängigen – Therapie mit Renin-Angiotensin-AldosteronSystem-Inhibitoren (RAASi) auftritt [8]. Als Reaktion darauf wird in der ärztlichen Praxis häufig die RAASi-Dosis reduziert oder die RAASi-Therapie beendet [9]. Dies wiederum führt zu einem erhöhten Hospitalisierungs- und Mortalitätsrisiko bei Risikopatienten mit chronischer Nierenkrankheit, Herzinsuffizienz und Diabetes mellitus [9]. Trotz des klinischen Bedarfs gab es lange kaum me-
Chronische Hyperkaliämie: Nationaler Konsensus empfiehlt moderne Kaliumbinder
dizinische Innovationen, die bei Hyperkaliämie eine schnelle, effektive, gut verträgliche und langfristige Kontrolle der Serumkaliumkonzentrationen ermöglichten. Der Nationale Konsensus verdeutlicht, dass moderne Kaliumbinder wie Natriumzirconiumcyclosilicat (Lokelma®) die leitliniengerechte Anwendung von RAASi unterstützen und zu einer langfristigen Kaliumkontrolle beitragen können [1].
Hohe Praxisrelevanz
Die Prävalenz der Hyperkaliämie, im Allgemeinen definiert als Serumkaliumkonzentrationen ≥5,5 mmol/L [10], beläuft sich in der Gesamtbevölkerung zwar auf lediglich 2 – 3 % [6]. Ihre hohe Praxisrelevanz zeigt sich jedoch vor allem darin, dass sie bei Risikopatienten mit Herzinsuffizienz (HF), chronischer Nierenkrankheit (CKD), Diabetes mellitus (DM) oder therapieresistenter Hypertonie (HTN) überdurchschnittlich häufig auftritt:
• bei bis zu 40 % der HF-Patienten mit reduzierter Ejektionsfraktion (HFrEF) unter Mineralokortikoid-Rezeptor-Antagonisten (MRA)-Therapie (NYHA-Klasse III oder IV mit LVEF <35 % unter 50 mg Spironolacton) [5]
• bei bis zu 40–50 % der CKD-Patienten, vor allem in fortgeschrittenen Stadien, mit DM oder unter RAASi-Therapie (HK definiert als Serumkalium >5,0 mmol/L, in einigen untersuchten Studien auch höher) [6]
• bei bis zu 20 % der Diabetiker (HK definiert als Serumkalium ≥5,5 mmol/L) [11]
• bei bis zu 17 % der Patienten mit therapieresistenter Hypertonie unter zusätzlicher MRA-Therapie (HK definiert als Serumkalium dauerhaft >5,5 mmol/L oder einmalig bestätigt ≥6,0 mmol/L) [7]
Bei den kardiorenalen Risikopatienten erhöht eine HK – trotz ihres per se häufig symptomarmen Verlaufs [12] – das Risiko für kardiovaskuläre Morbidität und Mortalität, insbesondere wenn die Betroffenen eine Therapie mit Renin-Angiotensin-Aldosteron-System-Inhibitoren (RAASi) erhalten [13, 14]. Aus diesem Grund empfiehlt der Nationale Konsensus in diesen Fällen eine konsequente Behandlung der HK mit dafür zugelassenen Wirkstoffen sowie eine engmaschige Überwachung der Kaliumwerte, um eine potenziell lebensbedrohliche Elektrolytentgleisung zu verhindern und die für die Patienten erforderliche RAASi-Therapie so hoch wie tolerabel halten zu können [1].
FORUM ELEKTROLYTSTÖRUNGEN
RAASi-Therapie
Inhibitoren des Renin-Angiotensin-Aldosteron-Systems (RAASi) regulieren den Flüssigkeits- und Elektrolythaushalt und werden beispielsweise bei Patienten mit Herz-Kreislauf-Erkrankungen und chronischer Nierenkrankheit u. a. zur Blutdrucksenkung eingesetzt. Zu den RAASi gehören AngiotensinConverting-Enzyme-Inhibitoren (ACEi), Angiotensin-Rezeptor-Blocker (ARB), Angiotensin-Rezeptor-Neprilysin-Inhibitoren (ARNI), Renin-Inhibitoren und Mineralokortikoid-Rezeptor-Antagonisten (MRA). Unter der Therapie mit RAASi kann sich eine Hyperkaliämie entwickeln. In der ärztlichen Praxis wird als Reaktion häufig die RAASi-Dosis reduziert oder abgesetzt. Dies wiederum kann jedoch zu einem erhöhten Mortalitätsrisiko bei Risikopatienten mit chronischer Herz- oder Niereninsuffizienz beitragen. Gemäß der aktuellen Leitlinie der Kidney Disease: Improving Global Outcomes (KDIGO) und der European Society of Cardiology (ESC)-Leitlinie sowie dem Nationalen Konsensus sollte dies vermieden und stattdessen der Kaliumspiegel kontrolliert sowie die optimale RAASi-Dosis beibehalten werden.
Strukturierte Therapie mit modernen Kaliumbindern statt kurzfristiger Korrektur
Ein praxisnaher Algorithmus im Nationalen Konsensus unterstützt die behandelnden Ärzte, bietet Orientierung und führt strukturiert durch Diagnostik und Therapie der HK. Dabei wird die zentrale Bedeutung der RAASi hervorgehoben und empfohlen, ein Absetzen oder eine Dosisreduktion nach Möglichkeit zu vermeiden. Denn Patienten, die die maximal angestrebte RAASi-Zieldosis einnehmen, haben eine bessere Prognose – sowohl bei CKD als auch HF und Diabetes mellitus [9]. Stattdessen sollten kaliumsenkende Maßnahmen vor einer Therapiereduktion angewendet werden [1]. „Der Einsatz moderner Kaliumbinder wie Natriumzirconiumcyclosilicat
ermöglicht es, die prognostisch relevante Versorgung mit RAASi bei Menschen mit Herzinsuffizienz, chronischer Nierenkrankheit, Diabetes mellitus oder einem therapieresistentem Hypertonus aufrechtzuerhalten oder sogar leitliniengerecht zu intensivieren“, erläuterte Prof. van der Giet. „Damit können wir eine langfristige Kaliumkontrolle erreichen und zugleich die notwendige RAASiExposition sicherstellen, was einen echten Paradigmenwechsel in der Behandlung der chronischen Hyperkaliämie bedeutet.“ In diesem Zusammenhang betonte van der Giet, dass die HK oft chronisch verläuft und daher auch in der Regel eine dauerhafte Intervention nötig macht [1]. Außerdem geht eine hyperkaliämische Episode mit erhöhter Wahrscheinlichkeit mit weiteren Episoden in kürzeren Intervallen einher [15].
Dr. Schleß stellte in ihrem Vortrag die internationale Real-World-Untersuchung TRACK vor [2]. Diese analysierte anhand der Daten von 1.330 erwachsenen Patienten, die von Juli 2022 bis Dezember 2024 erhoben wurden, die Therapieentscheidungen und deren Auswirkungen bei HK in den Fachbereichen Nephrologie, Kardiologie und Allgemeinmedizin. In Deutschland nahmen 230 Patienten teil, von denen 98,7 % mindestens eine Komorbidität und 48,7 % bereits bei Studienbeginn wiederkehrende HK-Episoden aufwiesen. Berücksichtigt wurden Patienten, die 21 Tage vor Studieneinschluss eine Serumkaliumkonzentration von >5,0 mmol/L aufwiesen. Primäre Endpunkte waren die Entscheidungen im HK-Management, deren Rationale und die erwartete Behandlungsdauer. Die Dauer bis zur Normokaliämie, die Häufigkeit rezidivierender HK sowie die Inanspruchnahme von Leistungen des Gesundheitssystems wurden als sekundäre Endpunkte erfasst. Die mediane Follow-Up-Zeit betrug 11,9 Monate.
Die Ergebnisse für die deutsche Kohorte zeigen, dass die Überwachung der Kaliumspiegel bei 63,5 % die primäre Managementstrategie war. Bei Studienbeginn erhielten 9,6 % keine Therapie, 11,3 % eine kaliumarme Ernährung. Leitlinienempfohlene Kaliumbinder kamen bei lediglich 3,9 % zum Einsatz. Die Managementstrategien blieben
20
FORUM ELEKTROLYTSTÖRUNGEN
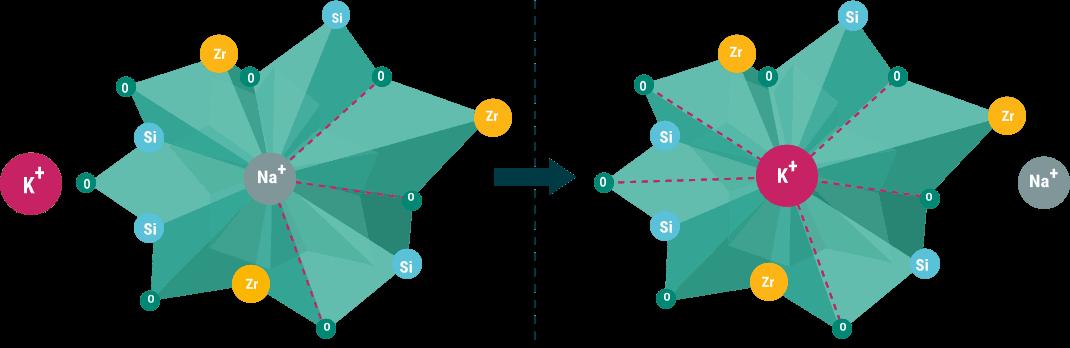
Natriumzirconiumcyclosilicat
Natriumzirconiumcyclosilicat (sodium zirconium cyclosilicate; SZC, Lokelma®) ist ein moderner hoch selektiver Kaliumbinder, der seit April 2021 in Deutschland zur Therapie der Hyperkaliämie bei Erwachsenen zur Verfügung steht [18]. Der Wirkeintritt erfolgt i. d. R. bereits nach einer Stunde, die mediane Zeit bis zum Eintreten einer Normokaliämie beträgt 2,2 Stunden (Interquartilbereich 1,0 bis 22,3 h). Die unter SZC erzielte Normokaliämie (definiert als mittleres
Serumkalium ≤5,1 mmol/L; 11-monatiges Follow-Up) konnte bei 88 % der Patienten langfristig aufrechterhalten wer den. Darüber hinaus setzten 89 % der Patienten unter SZC die Therapie mit Renin-Angiotensin-Aldosteron-System- In hibitoren (RAASi) nicht ab und 74 % konnten dieselbe Dosis beibehalten [18].
O: Sauerstoff; Si: Silicium; Zr: Zirconium
Natriumzirconiumcyclosilicat fungiert als eine nicht resorbierbare Kaliumfalle, die Kaliumionen (K +) im Austausch gegen Natriumionen (Na+) aufnimmt. O: Sauerstoff; Si: Silicium; Zr: Zirconium. Modifiziert nach Stavros et al. 8 Erhaltungstherapie: Chronische Hyperkaliämie langfristig behandeln
SZC besteht aus einem homogenen, mikroporösen Kristallgitter, an dem Protonen (H+) und Natriumionen (Na+) gebunden sind. Es wirkt im Magen-Darm-Lumen als selektive nicht resorbierbare Kaliumfalle, die Kaliumionen im Austausch mit Protonen und Natriumionen bindet. Infolge der vermehrten Ausscheidung von Kalium über die Fäzes reduziert sich die Kaliumkonzentration im Blutserum. SZC ist geschmacksneutral sowie geruchlos und kann ohne zeitlichen Abstand zu vielen Arzneimitteln oral angewendet werden. SZC wurde in mehreren klinischen Studien an 1.760 Patienten mit chronischer Nierenkrankheit (CKD), Herzinsuffizienz (HF) und Diabetes mellitus untersucht und im Allgemeinen gut vertragen [18].
medizinische Versorgung von Patient:innen mit Hyperkaliämie umfasst die langfristige Behandlung von mit chronischer Hyperkaliämie sowie die Notfalltherapie (s. u.).9 Die chronische Hyperkaliämie chronischen Erkrankungen wie einer chronischen Nierenkrankheit (CKD) , einer Herzinsuffizienz Diabetes mellitus (T2D) auf. 10
bei der überwiegenden Mehrheit (97,6 %) im Beobachtungszeitraum von 12 Monaten unverändert, und meist (62,0 %) wurde die Behandlung nur kurzfristig bis zum Erreichen der Normokaliämie fortgeführt [2].
Das HK-Management erfolgte auch nach einem Jahr meist (62,0 %) nur mittelfristig, d.h. bis zur Normalisierung der Kalium-
spiegel. 10 – 35 % der Studienteilnehmer, die eine Normokaliämie erreichten, waren 3 Monate danach bereits wieder von einer HK betroffen. Unter den 59 Patienten mit jeglicher therapeutischen Intervention erreichten 17 (28,8 %) das anvisierte Therapieziel nach einem Jahr nicht. Insgesamt erzielte etwa die Hälfte (53 %) zumindest einmal eine Normokaliämie und nach
einem Jahr lag bei 39,3 % weiterhin eine HK vor.
Diese Ergebnisse belegen eine hohe Rezidivrate, ohne eine stringente Behandlungsstrategie. Die Daten legen nahe, dass es in Deutschland ein nachhaltigeres Bewusstsein für das anhaltende HK-Risiko bei kardiorenalen Patienten braucht, um die HK langfristig zu monitorieren und zielgerichtet zu therapieren [2].
präventiven und therapeutischen Management der Hyperkaliämie wird beispielsweise CKD -Patient:innen die Kaliumaufnahme über die Nahrung zu reduzieren. 11 Eine kaliumarme Diät ist für die Betroffenen Hürden verbunden. Dies liegt einerseits daran, dass viele gängige Lebensmittel hohe Mengen Andererseits gelten zahlreiche kaliumreiche Nahrungsmittel wie verschiedene Obst - und Gemüsesorten „herzgesund“. 13
auf diese kann zur kardiovaskulären Krankheitslast der Patient:innen beitragen, die ohnehin
FORUM ELEKTROLYTSTÖRUNGEN
„Diese wiederkehrenden hyperkaliämischen Episoden sind nicht nur klinische Ereignisse, sondern ein Symptom struktureller Versorgungsdefizite. Solange die Strategien zum Langzeitmanagement, die uns zur Verfügung stehen, nicht in die Behandlungsrealität integriert werden, behandeln wir die Hyperkaliämie reaktiv statt präventiv –und das können wir besser.“ kommentierte Dr. Schleß.
PD Sven Waßmann riet zu folgendem Vorgehen: „Wir optimieren zunächst die RAASi-Therapie, überwachen die Kaliumspiegel und im Fall einer Hyperkaliämie behandeln wir diese leitliniengerecht – wenn erforderlich mit einem modernen Kaliumbinder. Das Ziel ist nicht nur, Kalium zu senken, sondern aus prognostischen Gründen die RAASi-Therapie langfristig aufrechtzuerhalten.“
Die Rationale erläuterte er anhand der retrospektiven Beobachtungsstudien ZORA-CKD [16] und ZORA-HF [17], die auf Daten der WIG2-Forschungsdatenbank mit über 4 Millionen gesetzlich Versicherten aus Deutschland (2014 bis 2022) basieren. Für die Auswertung wurden HK-Patienten mit
mindestens einem verschriebenen RAASi vor dem HK-Ereignis berücksichtigt. Die Betroffenen waren entweder an einer chronischen HF (ZORA-HF; n = 8.325) oder CKD (ZORA-CKD; n = 8.402) erkrankt. Abhängig vom RAASiManagement wurden sie der Gruppe „erhaltene RAASi-Therapie“ (Verschreibung/-en von mindestens derselben Anzahl an RAASi-Klassen wie vor dem HK-Indexereignis) oder der Gruppe „reduzierte RAASi-Therapie“ (Abbruch, d.h. keine Verschreibung für RAASi jeglicher Klasse, oder Abtitration, d.h. Verwendung von weniger RAASi-Klassen oder Dosisreduktion um ≥25 % in der Zeit nach dem HK-Indexereignis) zugewiesen, mittels Propensity-Score gematcht und die Behandlungsergebnisse über den Zeitraum von 12 Monaten ausgewertet. Die Behandlung mit RAASi wurde nach einer hyperkaliämischen Episode bei 46,5 % (ZORA-HF) bzw. 43,6 % (ZORA-CKD) reduziert. Patienten mit reduzierter RAASiDosis hatten ein höheres Risiko für Gesamthospitalisierungen jeder Ursache: Nach 12 Monaten lag dieses bei HF-Patienten bei 12 % (HR: 1,12; 95%-KI: 1,05–1,21, p < 0,001) und bei CKD-Patienten bei 9 % (HR: 1,09; 95%-KI: 1,02–1,17; p = 0,016) [16, 17].
Brigitte Söllner, Erlangen
Literatur
1 Eitel I et al. Dtsch Med Wochenschr 2026; 151: 221-228
2 Breitbart P et al. MMW-Fortschritte der Medizin 2025; 167(S5):1-24
3 Latus J et al. Hyperkaliämie bei Risikopatient:innen in deutschen Hausarztpraxen: Prävalenz, Diagnose und Therapie (WATCH-K-Studie). Abstract und Poster P15-04 zum 131. Kongress der Deutschen Gesellschaft für Innere Medizin e.V. 2025. https://doi.org/ 10.1007/s00108-025-01898-1.
4 ECC Committee, Subcommittees and Task Forces of the American Heart Association. Circulation 2005;112:IV1-203
5 Vardeny O et al. Circ Heart Fail 2014; 7:573-579
6 Kovesdy CP. Nat Rev Nephrol 2014; 10:653-662
7 Khosla N et al. Am J Nephrol 2009; 30:418-424
8 Betts KA et al. Curr Med Res Opin 2018;34:971-978
9 Epstein M et al. Am J Manag Care 2015;21:S212-S220
10 Zieschang, M AVP 2023;50:14-29
11 Haas JS et al. BMC Nephrol 2020;21: 332
12 Palmer BF et al. Nephrol Dial Transplant 2024;39:1097-1104
13 Collins AJ et al. Am J Nephrol 2017; 46:213-221
14 Hougen I et al. Kidney Int Rep 2021; 6:1309-1316
15 Rowan CG et al. Adv Ther 2024;41: 2381-2398
16 Latus J et al. Erhöhtes Hospitalisierungsrisiko bei CKD unter reduzierter RAASi-Therapie nach Hyperkaliämie: Aufschlüsse aus der ZORA-Studie. Abstract und Poster P24-07 zum 131. Kongress der Deutschen Gesellschaft für Innere Medizin e.V. 2025. https:// doi.org/10.1007/s00108-025-01898-1
17 Breitbart P et al. Decisions on RAASi therapy after hyperkalemia in heart failure – insights on hospitalizations from the ZORA study. Abstract und Poster #PP75 präsentiert am 26.08.2024 bei den DGK Herztagen, Hamburg. https:// doi.org/10.1007/s00392-024-02526-y
18 Fachinformation Lokelma®, aktueller Stand
22
Die hypertrophe-obstruktive Kardiomyopathie (HOCM) ist eine schwere, seltene und fortschreitende Erkrankung, die mit einer erheblichen Morbidität und Mortalität einhergeht. Ursache sind genetische Defekte in Sarkomerproteinen der Kardiomyozyten, die autosomal dominant vererbt werden. Frauen und Männer sind gleich häufig betroffen. Die Prävalenz liegt bei mindestens 1:500. Damit ist die HOCM die häufigste durch einen Erbgutfehler ausgelöste Herzerkrankung [1].
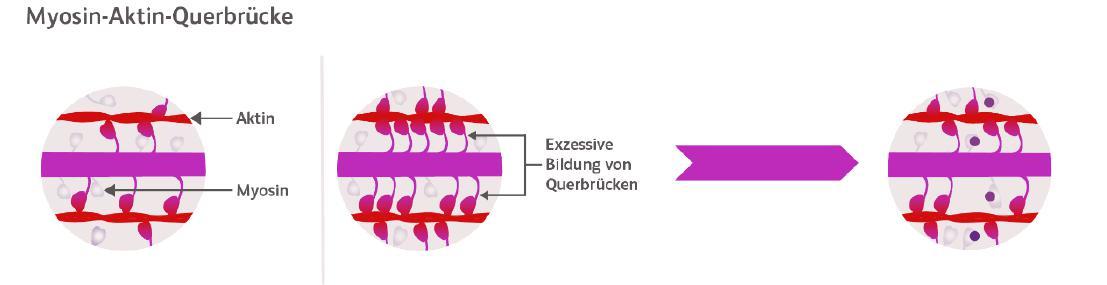
Charakteristisches Merkmal ist eine meist asymmetrische Hypertrophie des linken Ventrikels mit einer Wanddicke von ≥15 mm, die nicht durch systemische oder andere kardiale Erkrankungen wie arterielle Hypertonie oder Klappenerkrankungen zu erklären ist [2]. Die asymmetrische Verdickung des Herzmuskels verengt den linksventrikulären Ausflusstraktes (LVOT) und behindert damit – vor allem unter Belastung – den Blutausfluss aus der linken Kammer in die Aorta. Im Verlauf kommt es zusätzlich zur Obstruktion zu einer verminderten Relaxation und erhöhten Steifigkeit des kardialen Gewebes sowie zu einem erhöhten Energiebedarf des Herzmuskels [3]. Schwerwiegende Folge ist ein hohes Risiko für Vor-
FORUM CARDIOLOGICUM
SCOUT-HCM zeigt die Wirksamkeit von Mavacamten bei Jugendlichen mi t HOCM
hofflimmern, Schlaganfall, Herzinsuffizienz und einen plötzlichen Herztod [4].
Kardialer Myosin-Inhibitor greift gezielt in den Pathomechanismus der HOCM ein
Mavacamten (Camzyos®) ist der erste allosterische, reversible und selektive kardiale Myosin-Inhibitor, der ursächlich auf die zugrunde liegenden Pathomechanismen der HOCM abzielt (Abb. 1). Bei HOCM-Patienten führt die Inhibition des kardialen Myosins durch Mavacamten zu einer Normalisierung der Kontraktilität, einer Reduktion der dynamischen LVOT-Obstruktion und einer Verbesserung der kardialen Füllungsdrücke. Dies bestätigen die beiden zulassungsrelevanten Phase-IIIStudien EXPLORER-HCM und VALOR-HCM [5, 6], in denen der Myosin-Inhibitor klinisch bedeutsame und anhaltende Verbesserungen kardialer Parameter der chronischen Herzmuskelerkrankung erzielen konnte.
Aufgrund der überzeugenden Studienergebnisse wurde Mavacamten im Juni 2023 in allen Mitgliedsstaaten der Europäischen Union zur Behandlung der symptomatischen
hypertrophen obstruktiven Kardiomyopathie der NYHA-Klasse II–III bei erwachsenen Patienten zugelassen [7].
Potenzial auch für die Behandlung von Jugendlichen
Die Behandlungsmöglichkeiten für an HOCM erkrankte Jugendliche sind dagegen noch immer auf die medizinische Symptomkontrolle oder invasive Eingriffe beschränkt. Daher besteht weiterhin ein dringender Bedarf für eine kausale Therapie, zumal Patienten mit HCM unter 18 Jahren ein mehr als doppelt so hohes Risiko für einen plötzlichen Herztod wie Erwachsene zwischen 18 und 60 Jahren haben [8]. Außerdem beträgt die mediane Zeit bis zum Auftreten schwerwiegender kardialer Ereignisse oder der Notwendigkeit größerer kardialer Interventionen bei HOCM-Patienten im Kindesalter nur 1,5 Jahre ab Diagnosestellung [9]. Dies deutet auf ein enges Zeitfenster für krankheitsmodifizierende Interventionen hin.
Hoffnung machen nun die positiven Topline-Ergebnisse der von Bristol Myers Squibb initiierten Studie SCOUT-HCM, der ersten Phase-III-Studie, in der mit Ma-
die Wahrscheinlichkeit für die Bildung von kraftentwickelnden Querbrückenverbindungen während der Systole und der Enddiastole reduziert (bzw. bei HOCM normalisiert). 1
1 Mavacamten
FORUM CARDIOLOGICUM
die Wahrscheinlichkeit für die Bildung von kraftentwickelnden Querbrückenverbindungen während der Systole und der Enddiastole reduziert (bzw. bei HOCM normalisiert).
eine Verschiebung des Gesamtmyosins hin zu einem energiesparenden, aber superrelaxierten 1Zustand. Bei Patienten-HOCM führt eine Inhibition des durch Mavacamten zu einer Normalisierung der Kontraktilität, einer dynamischen Obstruktion-LVOT und einer Verbesserung der kardialen
die Wahrscheinlichkeit für die Bildung von kraftentwickelnden Querbrückenverbindungen während der Systole und der Enddiastole reduziert (bzw. bei HOCM normalisiert). 1 Mavacamten bewirkt ferner eine Verschiebung des Gesamtmyosins hin zu einem energiesparenden, aber rekrutierbaren, superrelaxierten Zustand.1 Bei HOCM-Patienten führt eine Inhibition des kardialen Myosins durch Mavacamten zu einer Normalisierung der Kontraktilität, einer Reduktion der dynamischen LVOT-Obstruktion und einer Verbesserung der kardialen Füllungsdrücke.1
bewirkt ferner eine Verschiebung des Gesamtmyosins hin zu einem energiesparenden, rekrutierbaren, superrelaxierten Zustand.1 Bei HOCM-Patienten führt eine Inhibition des kardialen Myosins durch Mavacamten zu einer Normalisierung der Kontraktilität, einer Reduktion der dynamischen LVOT-Obstruktion und einer Verbesserung der kardialen Füllungsdrücke.1
bewirkt ferner eine Verschiebung des Gesamtmyosins hin zu einem energiesparenden, rekrutierbaren, superrelaxierten Zustand.1 Bei HOCM-Patienten führt eine Inhibition kardialen Myosins durch Mavacamten zu einer Normalisierung der Kontraktilität, einer Reduktion der dynamischen LVOT-Obstruktion und einer Verbesserung der kardialen Füllungsdrücke.1
HOCM-Sarkomer mit Mavacamten (Normalisierung der Kontraktilität)
Abb. 1: Wirkmechanismus von Mavacamten.1, 2
Abb. 1: Wirkmechanismus von Mavacamten.1, 2
Abb. 1: Wirkmechanismus von Mavacamten.1, 2
Wirkmechanismus von 1,.Mavacamten 2
Abbildung 1: Kennzeichnend für die HOCM sind die übermäßige Bildung von Myosin-Aktin-Querbrücken und die Dysregulation des entspannten Zustands. Folgen sind eine kardiale Hyperkontraktilität, eine Hypertrophie und beeinträchtigte Relaxation des Herzmuskels sowie eine Obstruktion im LVOT und ein übermäßiger Energieverbrauch. Der selektive kardiale Myosin-Inhibitor Mavacamten reduziert die Anzahl der Myosinköpfchen im Sarkomer, die einen energiebereitstellenden Zustand erreichen können [3, 4]. So wird die Wahrscheinlichkeit für die Bildung von kraftentwickelnden Querbrückenverbindungen während der Systole und der Enddiastole reduziert (bzw. bei HOCM normalisiert). Außerdem bewirkt Mavacamten eine Verschiebung des Gesamtmyosins hin zu einem energiesparenden, aber rekrutierbaren, superrelaxierten Zustand [7].
Die zentrale Marktzulassung umfasst den Europäischen Wirtschaftsraum (EWR), d. h. Mavacamten ist zusätzlich zur Europäischen Union in Norwegen, Liechtenstein und Island zugelassen. 1
* Die zentrale Marktzulassung umfasst den Europäischen Wirtschaftsraum (EWR), d. h. Mavacamten ist zusätzlich Europäischen Union in Norwegen, Liechtenstein und Island zugelassen. 1
Marktzulassung umfasst den Europäischen Wirtschaftsraum (EWR), d. h. Mavacamten ist zusätzlich zur Norwegen, Liechtenstein und Island zugelassen.
* Die zentrale Marktzulassung umfasst den Europäischen Wirtschaftsraum (EWR), d. h. Mavacamten ist zusätzlich Europäischen Union in Norwegen, Liechtenstein und Island zugelassen.
vacamten ein Myosin-Inhibitor bei Jugendlichen mit symptomatischer obstruktiver hypertropher Kardiomyopathie untersucht wurde [10, 11]. In die randomisierte, doppelblinde, placebokontrollierte internationale Studie wurden 44 jugendliche Patienten (12 Jahre bis <18 Jahre) mit symptomatischer HOCM aufgenommen. Die Studie umfasst 3 Behandlungsphasen von insgesamt 200 Wochen: eine 28-wöchige placebokontrollierte Phase, gefolgt von einer 28-wöchigen Phase mit aktiver Behandlung (in der diejenigen Patienten, die auf Placebo randomisiert worden waren, zu Mavacamten wechselten) und eine offene Langzeitverlängerung (Long Term Extension) von bis zu 144 Wochen.
Primärer Endpunkt ist die Änderung vom Ausgangswert bis Woche
28 im Valsalva-LVOT-Gradienten.
Sekundäre Endpunkte umfassen Wirksamkeitsparameter wie den LVOT-Gradienten im Ruhezustand und nach Anstrengung, den maximalen Sauerstoffverbrauch, Erkrankungssymptome, den Gesundheitszustand der Patienten sowie Sicherheitsparameter und die Pharmakokinetik [10].
Die Studie erreichte den primären Endpunkt: Unter der Therapie mit Mavacamten zeigte sich nach 28 Wochen eine statistisch signifikante Reduktion im ValsalvaGradienten des linksventrikulären Ausflusstrakts (LVOT) gegenüber dem Ausgangswert im Vergleich zu Placebo [11]. Dies deutet auf die Wirksamkeit von Mavacamten bei der Verbesserung der LVOTObstruktion hin. Die statistische Signifikanz wurde ebenfalls in
mehreren sekundären Endpunkten erreicht, einschließlich der klinisch bedeutsamen Aspekte der Erkrankung.
Die Sicherheitsergebnisse stimmten mit dem etablierten Sicherheitsprofil von Mavacamten bei Erwachsenen überein. In der neuen, jüngeren Population wurden keine neuen Sicherheitssignale festgestellt.
Die positiven Studienergebnisse deuten darauf hin, dass Mavacamten das Potenzial haben könnte, als erster kardialer Myosin-Inhibitor zur Behandlung von Jugendlichen mit obstruktiver hypertropher Kardiomyopathie infrage zu kommen. Die Studie wird mit Phasen der aktiven Behandlung und der Langzeitverlängerung fortgesetzt [11].
Brigitte Söllner, Erlangen
Normales Sarkomer
HOCM-Sarkomer (Hyperkontraktilität)
24 FORUM CARDIOLOGICUM
Literatur
1 American College of Cardiology / European Society of Cardiology Clinical Expert: Consensus Document on Hypertrophic Cardiomyopathy. J Am Coll Cardiol 2003;42
2 Elliott PM et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 2014;35:2733-2779
3 Spudich JA. Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Pflugers Arch 2019; 471:701-717
4 Marian AJ et al. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res 2017;121:749-770
5 Olivotto I et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORERHCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020;396:759-769
6 Desai MY et al. Study design and rationale of VALOR-HCM: evaluation of mavacamten in adults with symptomatic obstructive hypertrophic cardiomyopathy who are eligible for septal reduction therapy. Am Heart J 2021;239:8089
8 Abdelfattah M et al. Temporal and global trends of the incidence of sudden cardiac death in hypertrophic cardiomyopathy. JACC Clin Electrophysiol 2022;8:1417-1427
9 Lafreniere-Roula M et al. Family screening for hypertrophic cardiomyopathy: is it time to change practice guidelines? Eur Heart J 2019;40: 36723681
10 Rossano J et al. Mavacamten in symptomatic adolescent patients with obstructive hypertrophic cardiomyopathy: design of the phase 3 SCOUT-HCM trial. Am Heart J 2026;292
11 Pressemitteilung vom 21.01.2026: Bristol Myers Squibb gibt positive ToplineErgebnisse der Phase-III-Studie SCOUT-HCM zur Bewertung von Mavacamten (Camzyos®) bei Jugendlichen mit symptomatischer obstruktiver hypertropher Kardiomyopathie (HOCM) bekannt.
Finerenon für Patienten mit
Herzinsuffizienz mit LVEF ≥40 % zur Zulassung in der EU empfohlen
Der Ausschuss für Humanarzneimittel (CHMP) der Europäischen Arzneimittel-Agentur (EMA) hat die Zulassung von Finerenon (Kerendia®) zur Behandlung von Erwachsenen mit Herzinsuffizienz und einer linksventrikulären Auswurfleistung (LVEF) ≥40 %, d.h. mit leicht reduzierter (HFmrEF) oder erhaltener (HFpEF) LVEF, empfohlen. Diese Patienten haben ein hohes Risiko für kardiovaskuläre Ereignisse, wobei Begleiterkrankungen wie chronische Nierenerkrankung, Bluthochdruck und Vorhofflimmern zu Krankenhausaufenthalten und erhöhter Sterblichkeit beitragen. Während bei HF mit einer LVEF ≤40 % Fortschritte in der Therapie erzielt wurden, gibt es für HF mit einer LVEF ≥40 % bislang nur begrenzte Behandlungsmöglichkeiten. Finerenon ist das erste Medikament, das auf den Mineralokortikoidrezeptor (MR)-Signalweg abzielt und die schädlichen Auswirkungen einer MR-Überaktivierung blockiert. Eine MR-Überaktivierung trägt zur Entwicklung einer chronischen Nierenerkrankung und zu kardiovaskulären Schäden bei, die durch metabolische, hämodynamische sowie entzündliche und fibrotische Faktoren verursacht werden können.
FINEARTS-HF zeigt signifikante und klinisch relevante kardiovaskuläre Vorteile
Die CHMP-Empfehlung basiert auf den positiven Ergebnissen der Phase-III-Studie FINEARTS-HF, in die 6.016 HF-Patienten mit einer LVEF ≥40 % (mittlere LVEF 53 ± 8 %) eingeschlossen wurden. Teilnahmevoraussetzungen waren eine strukturelle Herzerkrankung, eine kürzliche Hospitalisierung wegen Herzinsuffizienz oder erhöhte NTproBNP-Werte sowie eine Diuretika-Behandlung über mindestens 30 Tage vor der Randomisierung.
Teilnehmer und Studiendesign
Die Studienteilnehmer waren 72 ± 10 Jahre alt, 90 % hatten eine Hypertonie, 40 % einen Diabetes mellitus, 38 % Vorhofflimmern, 48 % eine eGFR <60 ml/ min/1,73m2; 69 % wurden der NYHA Klasse II zugeordnet. 85 % wurden mit Betablockern, 36 % mit ACE-Hemmern, 35 % mit ARB/ ARNI und 14 % mit SGLT2-Inhibitoren behandelt.
Zusätzlich zur üblichen Therapie von Symptomen und Komorbiditäten erhielten die Teilnehmer rando-
misiert entweder Finerenon (titriert bis 40 mg einmal tgl.) oder Placebo. Primärer Studienendpunkt war die Kombination aus kardiovaskulärem Tod und der Gesamtzahl (erstmalige und wiederkehrende) Herzinsuffizienz-Ereignisse, definiert als stationäre Aufnahmen oder Notfallbehandlungen aufgrund von Herzinsuffizienz.
Ergebnisse
Nach einer medianen Nachbeobachtungszeit von 32 Monaten hatte sich unter der Therapie mit Finerenon der kombinierte primäre Endpunkt im Vergleich zu Placebo signifikant vermindert (Rate Ratio: 0,84; 95%-KI: 0,74 – 0,95; p = 0,007): Der Anteil an kardiovaskulär bedingten Todesfällen betrug 8,1 % in der Finerenon- versus 8,7 % in der Placebo-Gruppe (Hazard Ratio: 0,93; 95%-KI, 0,78–1,11) und in der Finerenon-Gruppe kam es mit 842 gegenüber 1.024 Herzinsuffizienz-Ereignissen in der Placebo-Gruppe zu einer geringeren Verschlechterung der Herzinsuffizienz (Rate Ratio: 0,82; 95%KI: 0,71 – 0,94, p = 0,006).
FORUM CARDIOLOGICUM
Die im primären Endpunkt beobachteten Vorteile von Finerenon zeigten sich unabhängig von Hintergrundtherapie, Komorbiditäten oder Krankenhausaufenthaltsstatus und waren konsistent in allen vordefinierten Subgruppen, auch bei der Stratifizierung nach der Auswurfleistung oder der Vorbehandlung mit SGLT2-Inhibitoren [1].
Gutes Sicherheitsprofil
Ernste unerwünschte Ereignisse waren bei bei 38,7 % der Patienten in der Finerenon-Gruppe und bei 40,5 % in der Placebo-Gruppe zu verzeichnen. Erhöhungen des Kreatinins bzw. des Kalium-Spiegels waren häufiger unter Finerenon als unter Placebo, es gab aber keine fatale Episode einer Hyperkaliämie. Hypokaliämien waren seltener unter Finerenon als unter Placebo.
Fazit
Die Ergebnisse der Studie FINEARTS-HF zeigen, dass der nichtsteroidale MRA Finerenon bei Patienten mit Herzinsuffizienz und
leicht reduzierter oder erhaltener Ejektionsfraktion im Vergleich zu Placebo den kombinierten primären Endpunkt aus kardiovaskulärem Tod und der Gesamtzahl (erstmaligen und wiederkehrenden) Herzinsuffizienz-Ereignissen (definiert als Krankenhausaufenthalte oder dringende Arztbesuche aufgrund von Herzinsuffizienz) im Vergleich zu Placebo zusätzlich zur Standardtherapie signifikant reduzierte. Dabei zeichnet sich Finerenon durch ein gutes Sicherheitsprofil aus. Für die Patienten sind diese Ergebnisse hoch relevant, da es bislang neben der Gabe von SGLT2-Inhibitoren keine prognoseverbessernde Medikation für Betroffene mit HFpEF gibt. Hervorzuheben ist, dass es auch bei den mit SGLT2Inhibitoren vorbehandelten Studienteilnehmern zu einer ähnlichen Risikoreduktion kam wie bei den nicht vorbehandelten Patienten.
Brigitte Söllner, Erlangen
Quelle: Solomon SD. Finerenone in heart failure with mildly reduced or preserved ejection fraction. N Engl J Med 2024;391:1475-1485
Apixaban (Eliquis®) und Rivaroxaban (Xarelto®) sind die am häufigsten zur Behandlung der venösen Thromboembolie (VTE) bei Erwachsenen eingesetzten Nicht-Vitamin-K-abhängigen oralen Antikoagulanzien (NOACs) in Deutschland [1]. Ein direkter Vergleich der beiden NOACs in randomisierten kontrollierten Studien erfolgte bislang nicht, sondern nur ein Vergleich gegenüber Warfarin [2, 3, 4]. Für einen Vergleich zwischen den beiden NOACs lagen bisher lediglich Daten aus dem medizinischen Versorgungsalltag vor [5, 6], die zwar Assoziationen aufzeigen können, jedoch keine Kausalitäten.
Die aktuellen Daten der COBRRA (Comparison of Bleeding Risk Between Rivaroxaban and Apixaban for the Treatment of Acute Venous Thromboembolism)-Studie liefern nun erstmals Erkenntnisse aus einer randomisierten Studie, die den Einsatz von Apixaban und Rivaroxaban bei akuter VTE untersuchte [7].
Design und Zielsetzung der COBRRA-Studie
Die Investigator-initiierte, von der Pharmaindustrie unabhängige, pro-
FORUM ANTITHROMBOTICUM
COBRRA
– die erste Head-to-Head-
Studie der NOACs Apixaban und Rivaroxaban bei akuter venöser Thromboembolie
spektive, randomisierte, offene, Endpunkt-verblindete COBRRAStudie untersuchte 2760 erwachsene Patienten mit akuter symptomatischer Lungenembolie (LE) und/ oder proximaler tiefer Venenthrombose (TVT). Wichtige Einschlusskriterien waren eine bestätigte, neu diagnostizierte, symptomatische, akute VTE (TVT der proximalen unteren Extremitäten oder segmentale oder größere LE) sowie ein Alter ≥18 Jahre. Ausgeschlossen wurden Patienten mit >72 Stunden Antikoagulationstherapie, aktiven Blutungen, CrCl <30 ml/min, einer aktiven malignen Erkrankung, einem Gewicht >120 kg, einer Lebererkrankung (Child-Pugh-Klasse B oder C), mit einer anderen Indikation für eine Langzeitantikoagulation (z. B. Vorhofflimmern) sowie Frauen in Schwangerschaft oder Stillzeit [7].
Die Studienteilnehmer wurden 1:1 auf die Therapie mit Apixaban oder Rivaroxaban randomisiert (jeweils n = 1370) und erhielten über einen geplanten Behandlungszeitraum von 3 Monaten Apixaban 10 mg 2× täglich für 7 Tage, gefolgt von Apixaban 5 mg 2× täglich bzw. Rivaroxaban 15 mg 2× täglich für 21 Tage, gefolgt von Rivaroxaban 20 mg 1× täglich.
Primärer Endpunkt waren unabhängig beurteilte, adjustierte klinisch relevante Blutungen*. Wichtige sekundäre Endpunkte waren schwere Blutungen, klinisch relevante, nicht schwere (CRNM-)Blutungen, das Auftreten von VTE-Rezidiven sowie die Mortalität [7, 8].
Die Studie fand im Zeitraum vom 13. Dezember 2017 bis zum 30. April 2025 statt.
Über 50 %** geringeres Blutungsrisiko unter Apixaban bei vergleichbarer Wirksamkeit
Innerhalb von 3 Monaten trat bei 44 Patienten (3,3 %) des Apixaban-Studienarms sowie bei 97 Patienten (7,2 %) des Rivaroxaban-Studienarms eine klinisch relevante Blutung auf. Das entsprach einer relativen Risikoreduktion um etwa 50 % (Odds Ratio: 0,44; 95%-KI: 0,31 – 0,63; p < 0,00001) (Abb. 1).
VTE-Rezidive wurden unter Apixaban bei 15 Patienten (1,1 %) und unter Rivaroxaban bei 14 (1,0 %)
* Klinisch relevante Blutungen sind definiert als zusammengesetzter Endpunkt aus schweren Blutungen und klinisch relevanten nicht schweren Blutungen.
** Relative Risikoreduktion
FORUM ANTITHROMBOTICUM
Abbildung 1: Ergebnisse der COBRRA-Studie für den primären Endpunkt – klinisch relevante Blutungen* [7].
Abbildung 2: Ergebnisse der COBRRA-Studie für den sekundären Endpunkt – VTE-Rezidive [7].
• Sekundäre Endpunkte: VTE-Rezidive und Mortalität
Apixaban
VTE-Rezidive wurden unter Apixaban bei 15 Patient:innen (1,1 %) und unter Rivaroxaban bei 14 (1,0 %) beobachtet (OR 1,08; 95 %-KI: 0,52 – 2,25; p = 0,84 (Abb. 2)
Schwere Blutungen traten bei 5 Patient:innen (0,4 %) unter Apixaban und unter Rivaroxaban bei 31 Patient:innen (2,3 %) auf (OR 0,16; 95 %-KI: 0,06 – 0,41; p = < 0,00001).
Apixaban (Eliquis®) ist ein oraler, reversibler, hochselektiver, direkter Faktor Xa-Inhibitor. Die Substanz hemmt sowohl den freien als auch den im Prothrombinase-Komplex und im Thrombus gebundenen Gerinnungsfaktor Xa [10, 11]. Apixaban ist in der Europäischen Union in mehreren Indikationen zugelassen [12]. Basis hierfür sind Daten zur Wirksamkeit und Sicherheit, darunter Ergebnisse aus 7 Phase-III-Studien. Bei erwachsenen Patienten ist Apixaban zugelassen zur:
3 | 6
• Prophylaxe von Schlaganfällen und systemischen Embolien mit nicht-valvulärem Vorhofflimmern und einem oder mehreren Risikofaktoren wie Schlaganfall oder transitorischer ischämischer Attacke (TIA) in der Anamnese, Alter ≥75 Jahre, Hypertonie, Diabetes mellitus, symptomatischer Herzinsuffizienz (NYHA Klasse ≥II)
Die Mortalität lag im Apixaban-Studienarm bei 0,1 % und im Rivaroxaban-Arm bei 0,3 % (OR 0,25; 95 %-KI: 0,03 – 2,25)
• zur Prophylaxe venöser Thromboembolien (VTE) nach elektiven Hüft- oder Kniegelenkersatzoperationen
• zur Behandlung von tiefen Venenthrombosen (TVT) und Lungenembolien (LE) sowie
• zur Prophylaxe von rezidivierenden TVT und LE [12].
Schwere Blutungen traten bei 5 Patienten (0,4 %) unter Apixaban und bei 31 Patienten (2,3 %) unter Rivaroxaban auf (OR: 0,16; 95%-KI: 0,06 – 0,41; p = < 0,00001).
Die Mortalität lag im ApixabanStudienarm bei 0,1 % und im Rivaroxaban-Arm bei 0,3 % (OR: 0,25; 95%-KI: 0,03 – 2,25) [7].
Abb.
Fazit für die Praxis
In COBRRA, der ersten randomisierten, klinischen Head-to-HeadStudie zwischen Apixaban und Rivaroxaban, traten während einer dreimonatigen Behandlung von Patienten mit akuter symptomatischer VTE unter Apixaban signifikant weniger klinisch relevante Blutungen auf als unter Rivaroxaban [7]. Auch schwere Blutungen waren
unter Apixaban signifikant seltener – bei vergleichbarer Rate an VTERezidiven und vergleichbarer Mortalität [7].
Bereits zuvor lagen Daten aus dem Versorgungsalltag für Patienten mit akuter VTE vor, die Ärzten in der Praxis eine erste Orientierung bei der Wahl des geeigneten NOACs geben konnten [5, 6, 9] Die Daten der COBRRA-Studie zeigen nach Aussage der Autoren das
überlegene Sicherheitsprofil von Apixaban versus Rivaroxaban bei Patienten mit akuter VTE in einer unabhängigen, randomisierten klinischen Studie auf, sodass Ärzte in der täglichen Praxis künftig noch fundierter und sicherer zwischen den verfügbaren NOACs wählen können.
Brigitte Söllner, Erlangen
Sicherheit der Testosterontherapie von FDAExpertenpanel neu bewertet
Der männliche Testosteronmangel ist eine relevante, systemische Erkrankung. Hierzu präsentierte die US-amerikanische Food and Drug Administration (FDA) in einem Meeting klare wissenschaftliche Grundlagen. Bei entsprechender Indikation (männlicher Hypogonadismus) muss und darf eine adäquat dosierte Testosterontherapie (TTh) betroffenen Männern nicht länger aus Sorge vor Prostatakrebs oder kardiovaskulären Risiken vorenthalten werden.
Für Hausärzte sind die Ergebnisse des FDA-Expertentreffens vom 10.12.2025 zur TTh* von besonderer Bedeutung. Denn sie sind in der täglichen Praxis häufig die ersten Ansprechpartner für Männer mit
FORUM ANTITHROMBOTICUM
Literatur
1 IQVIA – IMS Health Retail (sell-out) & Hospital, Stand Juni 2025
2 Patel MR et al. N Engl J Med 2011; 365:883-891
3 Granger CB et al. N Engl J Med 2011; 365:981-992
4 Agnelli G et al. N Engl J Med 2013; 369:799-808 (+ Suppl.)
5 Dawwas GK et al. Ann Intern Med 2022;175:20-28
6 Bea S et al. JAMA Intern Med 2025; 185:837-846
KONGRESSE
unspezifischen, multifaktoriellen Beschwerden.
Symptome wie chronische Müdigkeit, Antriebslosigkeit, depressive Verstimmung, Libidoverlust, Muskelabbau oder Gewichtszunahme werden im hausärztlichen Setting oft isoliert oder symptomorientiert behandelt, oft ohne den zugrunde liegenden hormonellen Status zu erfassen.
7 Castellucci L. Präsentation auf dem ISTH-Kongress 2025, Late-Breakthrough Session 8023, 22. Juni 2025, Washington D.C., USA
8 https://clinicaltrials.gov/study/ NCT03266783
9 Glise Sandblad KG et al. J Intern Med 2023;294:743-760
10 Ansell J. J Thromb Haemost 2007;5 (Suppl 1):60-64
11 Anderson FA et al. Circulation 2003; 107(23 Suppl 1):9-16
12 Fachinformationen Eliquis® 5 mg, 2,5 mg; aktueller Stand
Keine Bedenken mehr hinsichtlich Prostatakarzinom oder kardiovaskulärer Ereignisse
Im Zentrum des FDA-Expertentreffens stand die Sicherheitsbewertung der TTh, insbesondere in Bezug auf kardiovaskuläre Ereignisse und das Prostatakarzinom (PCa) –zwei Aspekte, die in der ärztlichen Praxis bislang häufig zu einer unangebrachten Zurückhaltung hinsichtlich der TTh führen. Entscheidend für die Neubewertung sind insbesondere die Ergebnisse der TRAVERSE-Studie** aus dem Jahr 2023, die als bislang größte rando-
misierte, doppelblinde, placebokontrollierte Sicherheitsstudie zur TTh gilt. In diese Studie wurden mehr als 5.200 Männer mit symptomatischem Testosteronmangel eingeschlossen. Alle Patienten hatten ein erhöhtes kardiovaskuläres (CV) Risiko oder bereits eine CV-Erkrankung in der Vorgeschichte. Gerade diese Patientengruppe entspricht in hohem Maße der Realität hausärztlicher Versorgung. Die Ergebnisse zeigen, dass eine physiologische Testosterongabe weder mit vermehrten schweren unerwünschten CV-Ereignissen (Myokardinfarkt, Schlaganfall oder Tod durch CV-Ereignis) noch mit einer Zunahme der PCa-Inzidenz oder -Mortalität assoziiert ist. Innerhalb von 4 Jahren zeigten sich diesbezüglich keine signifikanten Unterschiede zwischen der Testosteron- und Placebo-Gruppe.
Dr. Abraham Morgentaler, Urologe an der Harvard Medical School, USA, brachte die Bedeutung dieser Daten auf den Punkt: Seiner Mei-
** Lincoff AM et al. Cardiovascular safety of testosterone-replacement therapy. N Engl J Med 2023;389:107-117
nung nach hat TRAVERSE die seit Jahren bestehenden großen Sicherheitsbedenken, nämlich die Angst vor Herz-Kreislauf-Risiken und Prostatakrebs, ausgeräumt.
„Nicht valider als der Glaube an die Zahnfee“
Ein weiterer zentraler Aspekt des Meetings war die Neubewertung des Zusammenhangs zwischen Testosteron und Prostata. Das jahrzehntelang gelehrte Dogma, Testosteron fördere das Wachstum oder die Progression eines PCa, wurde von mehreren Referenten als wissenschaftlich überholt dargestellt. Zahlreiche randomisierte Studien, Metaanalysen und populationsbasierte Registerdaten zeigen konsistent, dass weder endogen hohe Testosteronspiegel noch eine exogene, adäquat dosierte Testosterongabe das PCa-Risiko erhöhen. Im Gegenteil mehren sich die Hinweise darauf, dass niedrige Testosteronwerte mit aggressiveren PCa-Tumorformen und einer ungünstigeren Prognose assoziiert sein können. Morgentaler kommentierte diese Neubewertung: „Die Vorstellung, Testosteron sei gefährlich für die Prostata, ist heute wissenschaftlich nicht valider als der Glaube an die Zahnfee.“ Für die Hausärzte bedeutet das, dass hypogonadalen Männern eine indizierte TTh nicht aufgrund pauschaler Prostataängste vorenthalten werden sollte.
Label-Warnhinweise in den USA überholt
Großen Raum nahm im Meeting auch die Einordnung des Testos-
teronmangels als systemische Erkrankung ein. Niedrige Testosteronspiegel sind mit einer erhöhten Prävalenz von Typ-2-Diabetes, metabolischem Syndrom, Adipositas, Sarkopenie, Osteoporose, Depression sowie einer erhöhten Gesamtmortalität assoziiert. Mehrere Teilnehmer des Panels bezeichneten Testosteron als einen der wichtigsten verfügbaren Biomarker für die allgemeine Männergesundheit.
Dr. Helen Bernie, Urologin an der Indiana University, USA, brachte dies auf den Punkt: „Testosteron ist einer der aussagekräftigsten Biomarker für die Gesundheit des Mannes – vergleichbar mit Blutdruck oder HbA1c.“ Für die hausärztliche Versorgung folgt daraus, dass ein unbehandelter Testosteronmangel kein neutraler Befund ist, sondern ein potenziell modifizierbarer Risikofaktor mit langfristiger prognostischer Bedeutung. Bereits 2025 wurde auf Basis der TRAVERSE-Daten in den USA der kardiovaskuläre Warnhinweis im Label von Testosteron-Präparaten entfernt. Da eine TTh möglicherweise sogar dazu beitragen könnte die Entwicklung eines PCa zu verhindern, seien die Prostata-Kontraindikationen und Warnhinweise im Label ebenfalls überholt und sollten entfernt werden, befand das Panel einhellig.
Fazit
Das FDA-Expertentreffen lieferte für die hausärztliche Praxis eine klare Botschaft: Männer mit symptomatischem Testosteronmangel, die eine TTh erhalten, haben dadurch weder ein erhöhtes CV- noch PCaRisiko. Dies ist durch hochwertige
Evidenz, insbesondere durch die TRAVERSE-Studie, überzeugend belegt. Die Wirksamkeit einer TTh reicht weit über die Behandlung sexueller Symptome hinaus und umfasst zentrale Aspekte der metabolischen, muskuloskelettalen und psychischen Gesundheit.
Fabian Sandner, Nürnberg
Hämophilie A – was können wir in Bezug auf die Lebensqualität erreichen?
Hämophilie A ist gut behandelbar, doch was ich wichtig, damit die Patienten ein möglichst normales Leben ohne Ausgrenzung führen können? Dieser Frage gingen Experten anlässlich eines Symposiums während der diesjährigen Tagung der Gesellschaft für Thrombose- und Hämostaseforschung (GTH) nach. Professor Christoph Königs, Frankfurt, erläuterte die angestrebten Therapieziele: „Wir wollen ‚zero‘ Blutungen erreichen und zudem eine bessere Beweglichkeit, eine Erhaltung der Gelenkfunktion und eine Reduktion von Schmerzen für die Betroffenen ermöglichen“. Um das zu gewährleisten, gibt es allerdings in jeder Lebensphase neue therapeutische Herausforderungen. Laut PD Martin Olivieri, München, ist eine frühe Prävention bereits in den ersten Lebenstagen unter Einbeziehung der Familie für den weiteren Verlauf der Erkrankung wichtig. Hier gehe es darum, das Risiko für Hemmkörper und intrakraniale Blutungen zu reduzieren. Später im Erwachsenenalter sollte man auch das erhöhte Risiko für intrakrania-
le Blutungen im Auge behalten und möglichst niedrig halten, ebenso wie die weit verbreitete Schmerzsymptomatik. Und da die Betroffenen ob der guten Therapiemöglichkeiten immer älter werden, geht es auch darum, einer kardiovaskulären Problematik oder sonstigen altersbedingten operativen Eingriffen zu begegnen. So ist laut PD Katharina Holstein, Universitätsklinikum Schleswig-Holstein, beispielsweise das Schlaganfallrisiko genauso hoch wie bei der Normalbevölkerung. In allen Lebensabschnitten steht der Erhalt der Gelenkgesundheit im Vordergrund, darin waren sich die Referenten einig. Eine mögliche Behandlungsoption für einen solchen multimodalen Ansatz ist mit Efanesoctocog alfa (Altuvoct®) gegeben. Efanesoctocog alfa ist die einzige Therapie, die nachweislich die Obergrenze des von-WillebrandFaktors durchbricht, welche die Halbwertszeit der derzeitigen Faktor-VIII-(FVIII)-Therapien einschränkt. So lassen sich für bis zu 4 Tage nach Infusion normalisierte bis nahezu normale FaktorVIII-Aktivitätswerte von über 40 % erreichen. Königs dazu: „Die mediane Blutungsrate unter Efanesoctocog alfa liegt bei 0. Das ist ein enormer Fortschritt. Selbst bei bereits geschädigten Gelenken beobachten wir im Alltag eine verbesserte Funktion und deutlich weniger Schmerzen. Für die Betroffenen bedeutet das, keine blauen Flecken und Mut zu Aktivitäten im Alltag, die vorher stark eingeschränkt waren“.
Elke Engels, Bad Vilbel
Hämophilie A –konstanter
Blutungsschutz und mehr Lebensqualität unter Emicizumab
Hämophilie A (HA) wird zunehmend besser therapierbar, seitdem es gut untersuchte subkutane Therapieoptionen wie Emicizumab (Hemlibra®) gibt. Die Betroffenen profitieren im Vergleich zu einer intravenösen Darreichungsform von mehr Flexibilität und Lebensqualität.
Anlässlich der diesjährigen Jahrestagung der Gesellschaft für Thrombose und Hämostaseforschung (GTH) wurden die Vorteile der seit Jahren etablierten Emicizumab-Therapie auf Basis von Real-World-Evidenz-Erhebungen (RWE) vorgestellt.
Den Experten zufolge konnte eine signifikante Reduktion der Blutungshäufigkeiten dokumentiert werden, was die Teilhabe am Leben fördert, und die Therapiebelastung minimiert. Das sei wichtig, denn die Patienten sind krankheitsbedingt Außenseiter in Bezug auf soziale Kontakte, Ausbildungs- oder Berufsausübungsmöglichkeiten oder sportliche Kontakte. „Mit der Möglichkeit, Aktivitäten wie Sport auszuüben, steigt auch die Lebensqualität“, so Professor Charles Hay, Manchester. Er betonte, dass gerade Kinder mit Hämophilie A am Sport teilhaben wollen – sowohl in der Schule als auch auf dem Fußballplatz. Aktuelle Studien zeigen zudem eine nachhaltige Gelenkgesundheit unter der Therapie.
Hohe Wirksamkeit und Sicherheit auch bei Kindern
Hay präsentierte aktuelle Daten des britischen Registers UKHCDO, die die durchgehend hohe Wirksamkeit und Sicherheit von Emicizumab bei schwerer HA selbst bei vulnerablen Gruppen wie Säuglingen und Patienten mit Inhibitor-Historie belegen.
Ergänzend dazu unterstreicht die RWE aus dem PedNet-Register die Sicherheit und Wirksamkeit der Emicizumab-Prophylaxe bei Kindern mit HA. PD Katharina Holstein vom Universitätsklinikum Schleswig-Holstein zitierte dazu auch Daten der NIS EMIIL: Die annualisierte Blutungsrate (ABR) lag nach 2,5 Jahren bei 0,66. Hier konnte eine stabile Hämostase ohne Belastungsspitzen gezeigt werden – eine wichtige Basis für eine verbesserte Gelenkfunktion und eine aktivere Lebensgestaltung.
Ausblick mit NXT007 –Weiterentwicklung von Emicizumab
Spannende Einblicke in Bezug auf eine gezielte strukturelle Optimierung von Emicizumab gab Dr. Maria Elisa Manusco, Mailand: „Aufgrund der längeren Halbwertszeit sind längere Dosisintervalle möglich, wie die Phase-I- und -II-Studien zu NXT007 zeigen“. Die Studien ergaben hohe Raten an blutungsfreien Patienten (86,4 % unter Erhaltungstherapie) und eine niedrige ABR von 0,62
Kongresse / Mitteilungen
NXT007
NXT007 ist ein bispezifischer Antikörper der nächsten Generation zur Behandlung der Hämophilie A. Er wurde durch die Optimierung von Emicizumab entwickelt und ahmt die Kofaktorfunktion von aktiviertem Faktor VIII nach, um die intrinsische Blutgerinnung wiederherzustellen. Erste Studien zeigen sein Potenzial, die Thrombingenerierung zu steigern und die Hämostase auf das Niveau einer gesunden Person zu bringen.
NXT007 zeigt im Vergleich zu Emicizumab eine verbesserte Aktivität in präklinischen Tests:
• eine erhöhte Kofaktorfunktion durch optimierte leichte und schwere Ketten sowie
• eine längere Halbwertszeit durch eine angepasste konstante Antikörperregion (C-Terminus).
Sicherheit und Verträglichkeit: Im Rahmen einer globalen Phase-I/II-Studie wurden die Sicherheit und Verträglichkeit von NXT007 bei wiederholter Verabreichung steigender Dosen untersucht (Multiple-Ascending-Dose, MADStudie). Über durchschnittlich 68,4 Wochen wurden keine Sicherheitsbedenken festgestellt. In den beiden höchsten Dosiskohorten traten während der Erhaltungsdosis-Periode keine behandlungsbedürftigen Blutungen auf.
Hereditäres Angioödem: G-BA erkennt beträchtlichen Zusatznutzen für den FXIIa-Inhibitor Garadacimab
Ein Beschluss, der die Weichen der HAE-Behandlung neu stellt: Der Gemeinsame Bundesausschuss (GBA) hat dem Faktor-XIIa-Inhibitor Garadacimab (Andembry®) einen beträchtlichen Zusatznutzen in der Langzeitprophylaxe des hereditären Angioödems (HAE) attestiert. Im indirekten Vergleich zeigte Garadacimab signifikante Vorteile bei der Verbesserung von Attackenrate, Gesundheitszustand, Lebensqualität und Aktivitätsbeeinträchtigungen.
Monoklonaler IgG4-Antikörper mit innovativem Wirkmechanismus
(95%-KI: 0,3 – 1,0). Die Wiederherstellung der Thrombinbildung auf das Niveau gesunder Personen in den höchsten Kohorten weist der Expertin zufolge auf eine mögliche Hämostase-Normalisierung hin.
Elke Engels, Bad Vilbel
Das hereditäre Angioödem (HAE) ist geprägt von unvorhersehbaren, belastenden und teilweise lebensbedrohlichen Schwellungen der Haut und Schleimhäute – Symptome, die den Körper und die Lebensqualität deutlich beeinträchtigen. Entsprechend rücken die vollständige Krankheitskontrolle und die Normalisierung des Lebens als leitliniendefinierte Ziele zunehmend in den Fokus. Garadacimab (Andembry®), ein first-in-class, vollständig humaner, rekombinanter, monoklonaler IgG4-Antikörper gegen den aktivierten Faktor XII (FXIIa), ist seit Februar 2025 als Langzeitpro-
phylaxe (LTP) für HAE-Patienten ab 12 Jahren zugelassen. Durch die Hemmung der katalytischen Aktivität von FXIIa stoppt Garadacimab die Kallikrein-Kinin-Kaskade am Ursprung und verhindert dadurch die Bildung von HAE-typischen Angioödemen.
Die Effektivität des innovativen Wirkmechanismus wird durch die Studienergebnisse der 6-monatigen doppelblinden, randomisierten, kontrollierten Phase-III-Studie VANGUARD veranschaulicht: Der primäre Endpunkt wurde mit einer prüfärztlich bewerteten Reduzierung der monatlichen HAEAttackenrate um 87 % vs. Placebo (p < 0,0001) erreicht. Sekundäre Ergebnisse zeigten zudem: 62 % der Patienten blieben attackenfrei über den gesamten Beobachtungszeitraum; die Lebensqualität verbesserte sich signifikant; das Sicherheitsprofil war vergleichbar mit dem der Placebogruppe.
Begründung des Zusatznutzens
Richtungsweisend für die patientenorientierte Versorgung: Der G-BA hat im Rahmen der frühen Nutzenbewertung Garadacimab einen Anhaltspunkt für einen beträchtlichen Zusatznutzen zugesprochen. Diese Entscheidung gründet auf einem ausführlichen Dossier zu Garadacimab, bereitgestellt von CSL Behring und ausgewertet durch das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG), sowie Stellungnahmen von Experten im Anhörungsverfahren. In Abwesenheit von direkten Vergleichsstudien wurde ein sogenannter adjustierter indirekter Vergleich mit dem Plasma-Kalli-
krein-Inhibitor Berotralstat über den Brückenkomparator Placebo vorgenommen. Die VANGUARD-Studie für Garadacimab sowie die APeX-2und APeX-J-Studien für Berotralstat konnten dank ihrer starken Ähnlichkeiten bei Studiendesign und -dauer sowie der Probandenpopulation einander gegenübergestellt werden.
HAE-Attacken im Fokus
Die monatliche HAE-Attackenrate galt in allen analysierten Studien als primärer Endpunkt; die Attacken wurden über elektronische Patiententagebücher erfasst und prüfärztlich bzw. durch unabhängige Experten bestätigt. Im adjustierten indirekten Vergleich zeigte sich für Garadacimab gegenüber Berotralstat ein statistisch signifikanter Vorteil bei der Reduktion der monatlichen HAE-Attackenrate, der vom G-BA als Anhaltspunkt für einen beträchtlichen Zusatznutzen eingestuft wurde. Für die Attackenfreiheit wurde kein signifikanter Unterschied festgestellt.
Signifikant verbesserte Lebensqualität unter Garadacimab
HAE beeinträchtigt Patienten nicht nur akut während der Attacke, sondern wirkt sich auch langfristig auf Alltag und Lebensqualität aus. Viele Betroffene leben in ständiger Angst vor der nächsten Episode, was zu Einschränkungen bei Beruf, Freizeit und Sozialleben führt. Anhand validierter Erhebungsinstrumente wurde die Veränderung der Parameter „Lebensqualität“, „Aktivitätsbeeinträchtigung“ und „all-
gemeiner Gesundheitszustand“ von Studienbeginn bis Behandlungsende zwischen den beiden Therapien verglichen und in den Beschluss aufgenommen. In allen drei analysierten Bereichen erzielten Patienten unter Garadacimab signifikant bessere Ergebnisse, die der G-BA als Zusatznutzen einstufte.
S. M.
Elektrodenloser Herzschrittmacher LivIQ
Biotronik hat seine DX-Technologie erweitert und mit LivIQ den ersten elektrodenlosen Herzschrittmacher mit AV-synchronem Pacing auf Basis atrialer elektrischer Fernfeldsignale entwickelt. Ende Januar 2026 wurde das Schrittmachersystem LivIQ erstmals erfolgreich beim Menschen implantiert. Die Eingriffe erfolgten im Rahmen der BIO|CONCEPT.LivIQ-Studie, einer frühen klinischen Untersuchung zur ersten Bewertung der Sicherheit und Leistungsfähigkeit des Systems.
Dr. Paul Gould, Brisbane, sowie Dr. Stewart Healy und Dr. Emily Kotschet, Melbourne, führten die Implantationen durch. Die australischen Ärzte hoben insbesondere das hervorragende Handling des Systems während der Prozedur hervor. „Ich war äußerst beeindruckt, wie einfach sich der Implantationskatheter bedienen ließ“, schilderte Gould über seine ersten Erfahrungen. „Er ermöglicht eine sehr präzise und stabile Positionierung und bei Bedarf ein unkompliziertes Repositionieren.“ Auch Healy und
Kotschet lobten die gute Sichtbarkeit von Gerät und Implantationskatheter während des Eingriffs: „Biotronik hat sich hier viele Gedanken gemacht. Die Sichtbarkeit der Verankerungen und des Tethers ist ausgezeichnet. Das System ist intuitiv und gut zu handhaben.“
AV-synchrones Pacing über atriale Fernfeldsignale
LivIQ ist der weltweit erste intrakardiale Herzschrittmacher, der über atriale Fernfeldsignale ein AV-synchrones Pacing in Ruhe und unter Belastung mit nur einem einzigen implantierbaren kardialen elektronischen Gerät (CIED) ermöglicht. Die intuitive Bedienbarkeit und die hervorragende Röntgensichtbarkeit aller wichtigen Komponenten erleichtern die Implantation erheblich. Das System ist darauf ausgelegt, eine zuverlässige Frequenzanpassung bereitzustellen, ohne die Lebensdauer des Geräts zu beeinträchtigen.
Elektrodenlose Schrittmacher finden zunehmend Anwendung, da sie Komplikationen konventioneller transvenöser Systeme wie z.B. Probleme an den Elektroden oder ein erhöhtes Infektionsrisiko reduzieren können. Das LivIQSystem von Biotronik verbindet diese Vorteile mit zusätzlichen funktionalen Möglichkeiten und setzt damit einen neuen Maßstab für die elektrodenlose Schrittmachertechnologie – für die Bradykardietherapie ein wichtiger Schritt.
Biotronik wird in den kommenden Monaten die globale zulassungsrelevante Studie starten.
S. M.
IMPRESSUM
PERFUSION
OFFIZIELLES ORGAN DER DEUTSCHEN GESELLSCHAFT FÜR ARTERIOSKLEROSEFORSCHUNG
Herausgeber:
Univ.-Prof. Dr. Dr. Edzard Ernst, Emeritus Professor of Complementary Medicine, University of Exeter, Peninsula Medical School,Salmon Pool Lane, Exeter EX2 4SG, UK
Prof. Dr. med. Wolfgang Koenig Deutsches Herzzentrum München Technische Universität München Lazarettstraße 36 80636 München
Wissenschaftlicher Beirat:
Prof. Dr. med. T. von Arnim (Kardiologie), München
Prof. Dr. med. G. V. R. Born (Arterioskleroseforschung), London
Prof. Dr. med. C. Diehm (Angiologie), Karlsbad
Priv.-Doz. Dr. med. Dr. phil. C. Drosde (Kardiologie), Freiburg
Dr. med. J. Dyerberg MD, Ph. D. (Klin. Chemie), Aalborg Sygehus, Dänemark
Univ.-Prof. Dr. med. H. W. Eichstädt, (Kardiologie), Berlin
Doz. Dr. rer. nat. F.-D. Ernst (Hämorheologie), Dresden
Dr. med. J. Gehring (Kardiologie, Rehabilitation), München
Prof. Dr. med. J. D. Gruß (Gefäßchirurgie), Kassel
Prof. Dr. J. Harenberg (Hämostaseologie), Mannheim
Prof. Dr. med. L. Heilmann (Gynäkologie), Rüsselsheim
Prof. Dr. med. H. M. Hoffmeister (Kardiologie), Solingen
Prof. Dr. med. H. U. Janka (Diabetologie), München
Dr. med. J. Janzen MPhil (Pathologie), Bern, Schweiz
Prof. Dr. med. L. Kollár M.D., PhD (Gefäßchirurgie), Universität Pécs, Ungarn
Prof. Dr. med. M. Marshall (Phlebologie), Rottach Egern
Prof Dr. med. J. Matsubara (Chirurgie), Ishikawa, Japan
Prof. Dr. med. G. Mchedlishvilli (Mikrozirkulation), Tbilisi, Georgien
Prof. Dr. med. V. Mitrovic (Kardiologie, Klinische Pharmakologie), Bad Nauheim
Prof. Dr. med. H. Mörl (Angiologie), Mannheim
Prof. Dr. med. F. J. Neumann (Kardiologie), Bad Krozingen
Prof. Dr. med. K. L. Resch (Medizin-Statistik), Bad Elster
Prof. Dr. med. G. Rettig (Kardiologie), Homburg
PD Dr. med. Rainer Röttgen (Radiologie), Berlin
Prof. Dr. med. G. Schmid-Schönbein (Biomechanik), La Jolla, USA
Prof. Dr. med. H. Schmid-Schönbein (Physiologie), Aachen
Prof. Dr. med. A. Schrey (Pharmakologie), Düsseldorf
Prof. Dr. med. H. Sinzinger (Nuklearmedizin), Wien, Österreich
Prof. Dr. med. T. Störk (Kardiologie, Angiologie), Göppingen
Prof. Dr. med. I. Szirmai M.D. (Neurologie), Universität Budapest, Ungarn
Prof. Dr. med. G. Trübestein (Angiologie), Bonn
Prof. Dr. med. B. Tsinamdzvrishvili (Kardiologie, Hypertonie), Tbilisi, Georgien
Prof. Dr. med. W. Vanscheidt (Dermatologie), Freiburg
Prof. Dr. med. H. Weidemann (Kardiologie, Sozialmedizin), Bad Krozingen
Schriftleitung:
Univ.-Prof. Dr. Dr. Edzard Ernst, Emeritus Professor of Complementary Medicine, University of Exeter, Peninsula Medical School, Salmon Pool Lane, Exeter EX2 4SG, UK
E-Mail: Edzard.Ernst@pms.ac.uk
Tel: +44 (0) 1392 726029
Fax: +44 (0) 1392 421009
Die Zeitschrift erscheint 6-mal im Jahr; Jahresabonnement 27,–; Einzelheft 5,50, inklusive MwSt., zuzüglich Versandspesen. Der Abonnementpreis ist im voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag.
Geschäftsführerin:
Sibylle Michna Anschrift wie Verlag
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Werbung, Beratung, Verkauf: Sibylle Michna (verantwortlich) Anschrift wie Verlag
Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
Erfüllungsort: Puschendorf
Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Anmerkung der Redaktion: Zur besseren Lesbarkeit werden in der PERFUSION personenbezogene Bezeichnungen, die sich auf das männliche oder weibliche Geschlecht beziehen, grundsätzlich nur in der männlichen Form verwendet. Damit wird keine Diskriminierung des Geschlechts ausgedrückt.
Ein nationaler Konsensus aus interdisziplinären Expert:innen* fordert bei Patient:innen mit einem erhöhten Hyperkaliämie-Risiko (z. B. bei CKD, HI oder DM) einen Paradigmenwechsel in der Behandlung.1,2
Praktische Handlungsempfehlungen aus dem nationalen Konsensus1
Langfristige Therapie i. d. R. dauerhafte Intervention erforderlich1
Medikationen
Zusätzlicher Einsatz von modernen Kalium-bindenden Substanzen1
Korrigierbare Faktoren Medikationscheck und Ernährungsanamnese1,3 RAASi reduzieren/absetzen als letzte Option1,3 !
Internationale Leitlinien empfehlen den Einsatz von Kaliumbindern wie LOKELMA®, Diuretika und ggf. Bicarbonaten.3–5
HIER MEHR ZU LOKELMA® ERFAHREN!
ALLE EMPFEHLUNGEN DER EXPERT:INNEN AUF EINEN BLICK!
* Autor:innen: Prof. Dr. med. Ingo Eitel, Priv.-Doz. Dr. med. Insa Emrich, Prof. Dr. Dr. Stephan von Haehling, Dr. med. Birgit Schleß, Prof. Dr. med. Roland Schmitt, Prof. Dr. med. Frank Strutz, Priv.-Doz. Dr. med. Sven Wassmann, Prof. Prof. h.c. Dr. med. Markus van der Giet.
1 Eitel I et al. Paradigmenwechel in der Behandlung der chronischen Hyperkaliämie zur Optimierung der kardiorenalen Prognose. Deutsche Medizinische Wochenschrift 2026. doi 10.1055/a-2755-3785. Online ahead of print. (Abrufbar unter: https://www.thieme-connect.com/products/ejournals/abstract/10.1055/a-2755-3785). 2 Hunter RW, Bailey MA. Nephrol Dial Transplant. 2019; 34(Suppl 3): iii2–iii11. 3 KDIGO 2024. Clinical Practice Guideline for the Evaluation and Management of CKD. (Abrufbar unter: https://kdigo.org/wp-content/uploads/2024/03/KDIGO-2024-CKD-Guideline.pdf). 4 McDonagh TA et al. Eur Heart J. 2021; 42(36): 3599–3726 (inkl. Suppl.). 5 Mancia G et al. J Hypertens. 2023; 41(12): 1874–2071.
Lokelma® 5 g Pulver zur Herstellung einer Suspension zum Einnehmen / Lokelma® 10 g Pulver zur Herstellung einer Suspension zum Einnehmen | Wirkstoff: Natriumzirconiumhydrogencyclohexasilicat Hydrat (3:2:1:1:x). Verschreibungspflichtig. Zusammensetzung: Lokelma 5 g Pulver zur Herstellung einer Suspension zum Einnehmen: 1 Beutel enthält 5 g Natriumzirconiumhydrogencyclohexasilicat Hydrat (3:2:1:1:x). Lokelma 10 g Pulver zur Herstellung einer Suspension zum Einnehmen: 1 Beutel enthält 10 g Natriumzirconiumhydrogencyclohexasilicat Hydrat (3:2:1:1:x). Sonstige Bestandteile: keine. Anwendungsgebiet: Zur Behandlung einer Hyperkaliämie bei erwachsenen Patienten. Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff. Nebenwirkungen: Sehr häufig: Verschlechterung einer bereits bestehenden Herzinsuffizienz. Häufig: Hypokaliämie, Obstipation, Ereignisse im Zusammenhang mit Ödemen. Weitere Hinweise: siehe Fachinformation. Pharmazeutischer Unternehmer: AstraZeneca GmbH, Friesenweg 26, 22763 Hamburg, E-Mail: azinfo@astrazeneca.com, www.astrazeneca.de, Servicehotline für Produktanfragen: 0800 22 88 660. Stand: Juli 2025.