JAHRGANG 35
HEFT 1
Februar 2026

JAHRGANG 35
HEFT 1
Februar 2026
Brensocatib – der erste Wirkstoff zur Behandlung der Non-CF-Bronchiektasen-Erkrankung
Eosinophile Ösophagitis: Langzeit- und Real-World-Daten belegen Wirksamkeit der orodispersiblen Budesonid-Schmelztablette
Morbus Parkinson: OFF-Episoden ganzheitlich angehen durch frühen Einsatz von Ongentys® und Kynmobi®
Rezidiviertes und refraktäres Multiples Myelom: Elranatamab ermöglicht verlängerte
Therapiepausen ohne Wirksamkeitsverlust
Hitzewallungen mit Elinzanetant hormonfrei behandeln
Überzeugende Wirksamkeitsdaten für die RSV-Impfung mit Abrysvo®: Hoher Impfschutz auch in der 2. Saison
Nipocalimab – ein neuer FcRn-Blocker zur Therapie der generalisierten Myasthenia gravis
Remibrutinib verbessert Symptome, Lebensqualität und Schlaf bei chronischer spontaner Urtikaria
Pirtubrutinib erhält Zulassungserweiterung für die Therapie der rezidivierten oder refraktären chronisch lymphatischen Leukämie VERL AG PERFUSION
Triglyceride1,2
Wirksam senken und dauerhaft unter Kontrolle*
Pankreatitis1,2
~ 9 von 10 wurden verhindert**
Besseres Sicherheitsprofil1,3
Weniger unerwünschte Ereignisse als unter Placebo
Fertigpen
Einfach patientenfreundliche Anwendung
1x monatlich
DER ApoC3 Inhibitor
DER NÄCHSTEN GENERATION

Tryngolza® wird angewendet bei erwachsenen Patienten ergänzend zu einer Diät zur Behandlung des genetisch bestätigten Familiären Chylomikronämie-Syndroms (FCS).
* placebokorrigierte Triglycerid-Reduktion um 59,4 % nach 12 Monaten.
** 88 % weniger Pankreatitiden als unter Placebo, gepooltes Pankreatitis Ereignisraten-Verhältnis 0.12 (KI: 0.022, 0.656).
1. Fachinformation Tryngolza® (Olezarsen), Stand November 2025. 2. Stroes ESG et al. N Engl J Med. 2024; 390(19):1781-179.2. 3. Gaudet et al. Atheroscler Suppl 2010; 11: 55–6. Weitere Informationen zu Warnhinweisen und Vorsichtsmaßnahmen für die Anwendung, Wechselwirkungen mit anderen Arzneimitteln oder sonstige Wechselwirkungen; Fertilität, Schwangerschaft und Stillzeit, Nebenwirkungen und Gewöhnungseffekte entnehmen Sie bitte der veröffentlichten Fachinformation.
ApoC3, Apolipoprotein C3; ASO, Antisense-Oligonukleotid; FCS, Familiäres Chylomikronämie-Syndrom
Sobi und Tryngolza® sind Marken von Swedish Orphan Biovitrum AB (publ).
© 2026 Swedish Orphan Biovitrum AB (publ) – Alle Rechte vorbehalten.
Tryngolza® 80 mg
▼ Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung
Tryngolza 80 mg Injektionslösung im Fertigpen · Wirkstoff: Olezarsen · Zusammensetzung: Jeder Einzeldosis-Fertigpen enthält 80 mg Olezarsen (als Olezarsen-Natrium) in 0,8 ml Lösung. Jeder ml enthält 100 mg Olezarsen (als Olezarsen-Natrium) · Liste von sonstigen Bestandteilen: Natriumdihydrogenphosphat (E339), Dinatriumhydrogenphosphat (E339), Natriumchlorid, Wasser für Injektionszwecke, Natriumhydroxid (zur pH-Einstellung) (E524), Salzsäure (zur pH-Einstellung) (E507) · Anwendungsgebiete: Tryngolza wird angewendet bei erwachsenen Patienten ergänzend zu einer Diät zur Behandlung des genetisch bestätigten familiären Chylomikronämie-Syndroms (FCS) · Gegenanzeigen: Überempfindlichkeit gegen den Wirkstoff oder einen der sonstigen Bestandteile · Nebenwirkungen: Sehr häufig: Kopfschmerzen, Erbrechen, Arthralgie, Erythem an der Injektionsstelle; Häufig: Überempfindlichkeit, Myalgie, Hautverfärbung an der Injektionsstelle,
Bis ein neues Medikament zugelassen wird, sind umfangreiche Tests vorgeschrieben, in denen die „Wirksamkeit und Verträglichkeit“ eines Medikaments genau bestimmt werden. Wie allgemein bekannt, braucht man zur quantitativen Abschätzung, wie gut wirksam ein Präparat ist, zwei Gruppen, von denen in der Regel eine das Medikament bekommt und die andere ein Scheinmedikament, das genauso aussieht, schmeckt etc. Keiner der Beteiligten darf wissen, wer welche der beiden Varianten verabreicht bekommt (sog. doppelte Verblindung).
Die Wirksamkeit eines Medikaments zeichnet sich oft schon in Studien mit kleinen Gruppen deutlich ab – und das ist auch gut so. Würde man 1000 Teilnehmer brauchen, um (nur) durch statistische Berechnungen erkennen zu können, dass ein Medikament gegen Kopfschmerzen „wirksam“ ist, wäre der Vorteil so minimal, dass er für einen Menschen mit Kopfschmerz ziemlich irrelevant wäre… Methodisch weniger einfach ist der Nachweis der Verträglichkeit. Die Toxizität einer Substanz zu testen, also das Ausmaß, in dem ein Medikament schädliche oder giftige Wirkungen in einem Organismus hervorrufen kann (Gewebeschädigungen, Funktionsstörungen etc.), ist dabei noch die einfachere Übung. Hier gilt typischerweise der Spruch des Paracelsus (die Dosis macht das Gift), und genügend hohe Dosen führen ggf. sehr zuverlässig zu substanztypischen „Nebenwirkungen“. Das kann man im Labor (meist zunächst in Zell- oder Tierversuchen), systematisch und mit steigenden Dosen, recht schnell und effizient evaluieren. Demgegenüber gleicht die Prüfung der Verträglichkeit, also Untersuchungen auf Nebenwirkungen, die nicht unmittelbar toxischer Natur sind, oft der be-
rühmten Suche nach der Stecknadel im Heuhaufen. Ein klassisches Beispiel sind allergische Reaktionen, die eben oft nur bei einer ganz spezifischen Prädisposition auftreten. Für den Protonenpumpeninhibitor Omeprazol ist z.B. unter „seltene Nebenwirkungen“ (betreffen maximal 1 von 1.000 Behandelten) Haarausfall angegeben. Würde man eine Studie gegen Placebo machen, bräuchte man mindestens 30.000 Teilnehmer in jeder Gruppe, um zuverlässig ungefähr 10 Teilnehmer mit Haarausfall mehr in der Gruppe mit dem Medikament erwarten zu können. Und die würden wohl hoffnungslos in der Masse der Teilnehmer untergehen, bei denen sich im Laufe der Teilnahme Anzeichen eines Haarausfalls bemerkbar machen (insgesamt sind wohl bis zu 70 % der Männer und 40 % der Frauen betroffen, und damit immerhin mehrere Hundert im üblichen Zeitraum der Studienteilnahme). Umgekehrt würde man bei einer solchen Studie viele „Unpässlichkeiten“ in großer Zahl erfassen, die einfach im Leben häufig zu beobachten sind wie Schlafstörungen, Schwindel, Kopf- und Gliederschmerzen und vieles mehr. Je häufiger eine bestimmte medikamentös wirksame Substanz verordnet wird, umso größer ist die Gefahr, dass sich solche „Nebenwirkungen“ (auch) bei regelmäßiger Einnahme zufällig beobachten lassen. Seit die Protonenpumpeninhibitoren (PPI) in den 1980er Jahren die Therapie der Refluxkrankheit revolutionierten, stand der Verdacht im Raum, dass die

Prof.
Dr. med. Karl-Ludwig Resch
Langzeittherapie mit PPI das Risko für Magenkrebs erhöhen würde. Auch neuere systematische Reviews und Meta-Analysen nährten diesen Verdacht, so ein im letzten Jahr erschienener systematischer Review [1], der schlussfolgerte: „Es gibt starke epidemiologische Belege für einen Zusammenhang zwischen Protonenpumpenhemmern und Magenkrebs“. Die „epidemiologischen Belege“ sprachen für eine Verdoppelung des Risikos auf Magenkrebs, immerhin der fünfthäufigste Krebs. Allerdings wurde in dieser Publikation auch darauf eingegangen, dass verschiedene relevante Störfaktoren wie „umgekehrte Kausalität“ protopathiebedingte Verzerrung, die Verzögerungszeit sowie das Herausrechnen möglicher Effekte von Helicobacter pylori und seiner Eradikation die Ergebnisse verfälscht haben könnten.
Das scheint nun endgültig Gewissheit dank einer überzeugenden Registerstudie, die auf die Daten aller Einwohner Dänemarks, Finnlands, Islands Norwegens und Schwedens im Zeitraum von 1994 bis 2020 zurückgreifen konnte [2]. Die Wissenschaftler identifizierten 17.232 Fälle von Magenkrebs und stellten jedem dieser Fälle 10 Vergleichspersonen ohne die Erkrankung gegenüber, die jeweils in Bezug auf Alter, Geschlecht und Nationalität vergleichbar („gematcht“) waren, also insgesamt 172.297 Vergleichspersonen. Fazit: „Die Ergebnisse zeigen keinen verbleibenden Zusammenhang zwischen der Langzeitanwendung von Protonenpumpenhemmern und dem Risiko eines MagenAdenokarzinoms.“
Wenn das bei allen schlechten Nachrichten, die das neue Jahr bereits gebracht hat, nicht mal eine wirklich gute ist!
Karl-Ludwig Resch, Nürnberg
Brensocatib – der erste Wirkstoff zur Behandlung der Non-CF-Bronchiektasen-Erkrankung 4 Brigitte Söllner
Eosinophile Ösophagitis: Langzeit- und Real-World-Daten belegen Wirksamkeit der orodispersiblen BudesonidSchmelztablette 8
Morbus Parkinson: OFF-Episoden ganzheitlich angehen durch frühen Einsatz von Ongentys® und Kynmobi® 10
Rezidiviertes und refraktäres Multiples Myelom: Elranatamab ermöglicht verlängerte Therapiepausen ohne Wirksamkeitsverlust
Hitzewallungen mit Elinzanetant hormonfrei behandeln 14
Überzeugende Wirksamkeitsdaten für die RSV-Impfung mit Abrysvo®: Hoher Impfschutz auch in der 2. Saison 17
Nipocalimab – ein neuer FcRn-Blocker zur Therapie der generalisierten Myasthenia gravis 19
Quellen
1 Brusselaers N, Khodir Kamal H, Graham D, Engstrand L. Proton pump inhibitors and the risk of gastric cancer: a systematic review, evidence synthesis and life course epidemiology perspective. BMJ Open Gastroenterol 2025 Apr 19;12(1):e001719. PMID: 40253055
2 Duru O, Santoni G, Holmberg D, Birgisson H, Kauppila JH, von EulerChelpin M, Ness-Jensen E, Lagergren J. Long term use of proton pump inhibitors and risk of stomach cancer: population based case-control study in five Nordic countries. BMJ 2026 Jan 21;392:e086384. PMID: 41565320
Remibrutinib verbessert Symptome, Lebensqualität und Schlaf bei chronischer spontaner Urtikaria 23
Pirtubrutinib erhält Zulassungserweiterung für die Therapie der rezidivierten oder refraktären chronisch lymphatischen Leukämie 25
Wissenswertes 16, 18, 22, 26, 28 Kongresse 28

VERTRAUEN SIE ÜBER ZEHN JAHREN ERFAHRUNG 1 :
HEMLIBRA ® die erste subkutane Dauerprophylaxe für Patient:innen mit Hämophilie A *2 die einzige Antikörpertherapie für Hämophilie A mit Langzeitdaten 3-5
*mit schwerer oder mittelschwerer (mit schwerem Blutungsphänotyp) Hämophilie A (hereditärer Faktor-VIII-Mangel) in jeder Altersklasse unabhängig vom Faktor-VIII-Hemmkörper-Status2
Referenzen
1. Uchida N et al. Blood. 2016 Mar 31;127(13):1633-41.
2. Fachinformation HEMLIBRA® , https://go.roche.de/Fachinformation Hemlibra.
3. Shima, M. et al. Hemophilia 2021;27(1):81-8
4. Mahlangu J et al. Res Pract Thromb Haemost. 2024;8(2):102364.
5. Callaghan MU et al. Blood. 2021;137(16):2231-2242.
Februar 2026. M-DE-00029419.
Zu den Pflichtangaben: https://go.roche.de/Hemlibra_PA
Hier mehr erfahren https://go.roche.de/ HemlibraErfahrung
Die nicht durch eine Mukoviszidose (zystische Fibrose, CF) bedingte Non-CF-Bronchiektasen-Erkrankung ist nach Asthma und COPD die dritthäufigste chronische Erkrankung der Atemwege [1]. In Deutschland leben derzeit etwa 150.000 Menschen damit [2]. Die Prävalenz der Bronchiektasen-Erkrankung nimmt sowohl hierzulande als auch weltweit zu [3, 4], dennoch bleibt sie eine der am meisten vernachlässigten Krankheiten in der Atemwegsmedizin [5].
Eine chronische Lungenerkrankung mit lebensbedrohlichen Atemwegsentzündungen
Kennzeichnend für die Non-CFBronchiektasen-Erkrankung sind die dauerhafte Erweiterung und abnormale Verdickung der Atemwege [3]. In diesen Aussackungen der Bronchien sammelt sich zäher Bronchialschleim, der sich nur schwer abhusten lässt [6]. Folge des Sekretstaus ist eine chronische Atemwegsentzündung, die durch Bakterien und andere Erreger, die sich im Sekret ansiedeln, noch verstärkt wird. Chronische Atemwegsinfektionen, wiederkehrende, insbesondere neutrophile Entzündungen, und eine beeinträchtigte mukoziliäre Clearance verstärken sich gegenseitig und führen letztlich zu irreversiblen Schäden am Lungengewebe [7, 8]. Dieser Circulus vitiosus wird zusätzlich vorangetrieben durch das Eingreifen neutrophiler Granulozyten, die zur Abwehr von bakteriellen Infektionen ausgeschüttet werden. Dabei setzen sie Serinproteasen frei, die in einer überschießenden Reaktion nicht nur die Krankheitserreger in den Atemwegen unschädlich ma-
Brigitte Söllner,
Erlangen
chen, sondern auch die Bronchialwände und das umliegende Lungengewebe angreifen. Die Erkrankung kann verschiedene Ursachen haben. In seltenen Fällen ist sie angeboren, häufiger entwickelt sie sich als Folge von schweren Atemwegsinfektionen oder Atemwegserkrankungen wie COPD und Asthma. Auch Autoimmunerkrankungen kommen als Ursache in Betracht. In ca. 40 % der Fälle bleibt die Ursache jedoch unklar [7].
Typische Symptome sind anhaltender Husten, eine übermäßige Sputumbildung, Kurzatmigkeit, wiederholte Infekte und Husten mit Blutbeimengungen [7, 9].
Bei mehr als 30 % der Patienten kommt es zu wiederholten Infektionen und Exazerbationen, die 2 – 4 Wochen andauern können und eine Antibiotikatherapie erfordern [10]. Die Exazerbationen sind assoziiert mit:
• einer verminderten Lebensqualität
• einer Verschlechterung der Lungenfunktion
• einem erhöhten Risiko für weitere Exazerbationen
• Krankenhausaufenthalten und
• einer erhöhten Mortalität [11]. Die Symptome beeinträchtigen die Lebensqualität der Betroffenen erheblich und führen zu einer signifikanten Einschränkung alltäglicher, beruflicher und sozialer
Aktivitäten [12]. Die Furcht vor einer Infektion oder einer Verschlimmerung der Symptomatik kann zudem zu sozialer Isolation führen, was Symptome wie Angst und Depression nach sich zieht, wovon 20 – 40 % der Patienten berichten [13].
Bisherige Behandlungsmöglichkeiten wie eine physiotherapeutische Atemtherapie, schleimlösende Mittel und Antibiotika zur Behandlung der Infektionen lindern zwar die Symptome, können die chronische Entzündung und die fortschreitende, irreversible Schädigung des Lungengewebes jedoch nicht stoppen, sodass als als letzte Therapiemöglichkeit nur eine Lungentransplantation bleibt [14].
DPP-1-Inhibitor Brensocatib greift gezielt in den entzündlichen Prozess ein
Mit Brensocatib (Brinsupri®) steht seit November 2025 in der EU der bislang erste und einzige spezifisch für die Behandlung der nicht durch zystische Fibrose (Non-CF, Mukoviszidose) bedingten Bronchiektasen zugelassene Wirkstoff zur Verfügung. Brensocatib ist der erste Dipeptidylpeptidase-1 (DPP-1)Inhibitor für den gezielten Einsatz gegen neutrophile Entzündungen, die als Treiber der entzündlichen Prozesse bei der Non-CF-Bron-
chiektasen-Erkrankung gelten. Zugelassen ist Brensocatib für die Behandlung von Patienten ab 12 Jahren mit zwei oder mehr Exazerbationen in den vorangegangenen 12 Monaten [15]. Das Arzneimittel wurde aufgrund des ungedeckten Therapiebedarfs im Rahmen des Priority-Medicine-Schemes (PRIME) beschleunigt zugelassen [16]. Die Zulassung durch die Europäische Kommission basiert auf den positiven Daten der Phase-III-Studie ASPEN [17] und der Phase-IIStudie WILLOW [18] und bezieht sich ausschließlich auf die 25 mgDosierung von Brensocatib.
Klinisch relevante Verbesserungen in der ASPEN-Studie
Die randomisierte, doppelblinde, placebokontrollierte Phase-IIIParallelgruppenstudie ASPEN ist die größte klinische Studie, die jemals zur Behandlung der NonCF-Bronchiektasen-Erkrankung durchgeführt wurde [17]. Eingeschlossen wurden 1.721 Patienten ab 12 Jahren (1.680 Erwachsene und 41 Jugendliche) mit Non-CFBronchiektasen-Erkrankung. Die Studienteilnehmer erhielten randomisiert eine von 2 Dosierungen von Brensocatib (25 mg: n = 575 bzw. 10 mg: n = 583) oder Placebo (n = 563) einmal täglich über 52 Wochen.
Primärer Endpunkt war die Reduktion der jährlichen Rate pulmonaler Exazerbationen während des 52-wöchigen Behandlungszeitraums. Pulmonale Exazerbationen waren definiert als Verschlechterung von mindestens 3 Leitsymptomen innerhalb von 48 Stunden mit zunehmendem Husten, erhöhtem Auswurfvolumen, eitrigem Auswurf oder verstärkter Dyspnoe oder verminderter Belastungsto-
Brensocatib (Brinsupri®) ist ein kompetitiver und reversibler Inhibitor der Dipeptidylpeptidase 1 (DPP-1). DPP1 aktiviert proinflammatorische neutrophile Serinproteasen (NSP) wie die neutrophile Elastase (NE) und die Proteinase 3 (PR3) während der Neutrophilenreifung im Knochenmark. Diese Enzyme werden von neutrophilen Granulozyten ausgeschüttet, die bei entzündlichen Prozessen zur Abwehr von bakteriellen Infektionen ins Gewebe einwandern. Im Zuge der Immunabwehr kommt es bei chronischen Atemwegsentzündungen wie der Bronchiektasen-Erkrankung jedoch oft zu einer überschießenden Reaktion und es werden zu viele Serinproteasen in die Atemwege freigesetzt. Diese machen dann nicht nur die Erreger unschädlich, sondern greifen auch die Bronchialwände sowie das umliegende Lungengewebe an und zählen damit zu den Haupttreibern der chronischen Atemwegsentzündung bei der Bronchiektasen-Erkrankung.
Brensocatib kann diesen Circulus vitiosus unterbrechen, indem es die Dipeptidylpeptidase 1 blockiert und damit die Aktivierung der proinflammatorischen neutrophilen Serinproteasen unterbindet. Diese Reaktion ist reversibel – wird Brensocatib abgesetzt, blockiert es DDP-1 nicht länger, sodass dieses die Enzyme wieder in vollem Umfang aktivieren und die Bakterienabwehr verstärken kann [15].
leranz und Fatigue und/oder Unwohlsein und Bluthusten, die eine ärztliche Verordnung systemisch wirksamer Antibiotika notwendig machten. Pulmonale Exazerbationen wurden als schwerwiegend eingestuft, wenn sie eine Behandlung mit intravenösen Antibiotika erforderten und/oder zu einem stationären Krankenhausaufenthalt führten.
Sekundäre Endpunkte waren die Zeit bis zur ersten Exazerbation innerhalb der 52 Wochen, der Anteil der Patienten, die in Woche 52 exazerbationsfrei blieben, die Veränderung des forcierten Einsekundenvolumens (FEV1 ) und die Veränderung der Lebensqualität [17].
Verringerung der jährlichen Rate pulmonaler Exazerbationen
Der primäre Endpunkt wurde erreicht. Beide untersuchten Dosie-
rungsstärken (10 mg, 25 mg) von Brensocatib erzielten bei einmal täglicher Gabe eine statistisch signifikante und klinisch relevante Verringerung der jährlichen Rate pulmonaler Exazerbationen während des 52-wöchigen Behandlungszeitraums im Vergleich zu Placebo. Die annualisierte Rate an pulmonalen Exazerbationen betrug 1,02 in der 10-mg-BrensocatibGruppe, 1,04 in der 25-mg-Brensocatib-Gruppe und 1,29 in der Placebo-Gruppe (Tab. 1) [17].
Signifikant weniger und weniger schwerer Exazerbationen sowie eine langsamere Verschlechterung der Lungenfunktion
Auch bei den sekundären Endpunkten war Brensocatib der Placebobehandlung überlegen: Die Zeit bis zur ersten Exazerbation war unter der Behandlung mit
Endpunkt
Primärer Endpunkt
Jährliche Rate pulmonaler Exazerbationen (Anzahl der Ereignisse pro Jahr)
Rate Ratio
Brensocatib 10 mg (n=583)
(95% KI: 0,91–1,13)
(95%-KI: 0,68–0,92)
Brensocatib
(95%-KI: 0,93–1,16)
(95%-KI: 0,69–0,94)
Adjustierter p-Wert 0,004 0,005
Sekundäre Endpunkte
Hazard-Ratio für die Zeit bis zur ersten Exazerbation
(95%-KI: 0,70–0,95)
(95%-KI: 0,70–0,97)
Adjustierter p-Wert 0,02 0,04
Anteil der Patienten, die während der Behandlung exazerbationsfrei blieben
Rate Ratio für das Ausbleiben der Exazerbation in Woche 52
Adjustierter p-Wert
FEV1-Wert nach Bronchodilatation
Mittlere Veränderung gegenüber dem Ausgangswert bis Woche 52
Mittlere Differenz zwischen Brensocatib und Placebo
(95%-KI: 1,06–1,37)
(95%-KI: 1,04–1,34)
(95%-KI: 1,16–1,43)
ml (95%-KI: −14 bis 37)
Adjustierter p-Wert 0,38
Jährliche Rate schwerer Exazerbationen (Anzahl der Ereignisse pro Jahr)
(95%-KI: 0,10–0,18)
Rate Ratio 0,74 (95%-KI: 0,51–1,09)
(95%-KI: 11–65)
(95%-KI: 0,11–0,18)
(95%-KI: 0,52–1,06)
Adjustierter p-Wert nicht angegeben 0,21
Lebensqualität (QOL-B RSS)
Mittlere Veränderung vom Ausgangswert bis Woche 52
6,84±0,77 Punkte
8,58±0,76 Punkte
Mittlere Differenz gegenüber Placebo 2,03 (95%-KI: –0,08–4,14 3,77 (95%-KI: 1,58–5,85)
(95 %-KI: 0,14–0,24)
4,81±0,75 Punkte
Tabelle 1: Ergebnisse der ASPEN-Studie. In allen Studienendpunkten war die Behandlung der Non-CF-Bronchiektasen-Erkrankung mit dem Dipeptidylpeptidase-1 (DPP-1)-Inhibitor der Placebo-Behandlung überlegen [17].
Brensocatib signifikant länger (Reduktion der Inzidenz der ersten Exazerbation unter 10 mg um 19 % und unter 25 mg um 17 % über den 52-wöchigen Zeitraum im Vergleich zu Placebo). Auch der Anteil der Patienten ohne Exazerbation war mit jeweils 48,5 % höher unter der 10-mg- und der 25-mg-Dosis Brensocatib als unter Placebo mit 40,3 %. Bei den
Patienten, die Brensocatib erhielten, verschlechterte sich die Lungenfunktion (gemessen als FEV1) signifikant langsamer – in Woche 52 war der FEV1-Wert unter der 10-mg-Dosis um 50 ml, unter der 25-mg-Dosis um 24 ml und unter Placebo um 62 ml gesunken. Auch hinsichtlich der jährlichen Rate an schweren Exazerbationen war Brensocatib überlegen: Unter
den beiden Brensocatib-Dosen betrug sie 0,14, unter Placebo 0,19 [17].
Diese Ergebnisse zeigen, dass die Hemmung von DPP-1 mit Brensocatib, insbesondere in einer Dosierung von 25 mg/d, und damit die Bekämpfung der neutrophilen Entzündung zu einer signifikanten Verbesserung der Behandlungsergebnisse bei Patienten mit Bron-
chiektasen führt, was sich auch positiv auf die Lebensqualität auswirkt (Tab. 1).
Die Behandlung mit Brensocatib wurde von den Patienten gut vertragen [1]. Die Häufigkeit von unerwünschten Ereignissen insgesamt sowie von unerwünschten Ereignissen, die zum Abbruch der Behandlung mit Brensocatib oder Placebo oder zum Ausscheiden aus der Studie führten, war in allen Gruppen vergleichbar. Die häufigsten unerwünschten Ereignisse, die unter beiden Brensocatib-Dosierungen häufiger auftraten als unter Placebo, waren COVID-19 (15,8 %, 20,9 %, 15,8 %), Nasopharyngitis (7,7 %, 6,3 %, 7,6 %), Husten (7,0 %, 6,1 %, 6,4 %) und Kopfschmerzen (6,7 %, 8,5 % und 6,9 %) – jeweils für Brensocatib 10 mg, Brensocatib 25 mg bzw. Placebo [1].
Schwere unerwünschte Ereignisse wurden bei 101 Patienten (17,4 %) in der 10-mg-Brensocatib-Gruppe, bei 97 Patienten (16,9 %) in der 25-mg-Brensocatib-Gruppe und bei 108 Patienten (19,2 %) in der Placebo-Gruppe berichtet. Todesfälle aufgrund unerwünschter Ereignisse traten bei 3 Patienten (0,5 %) in der 10-mg-Brensocatib-Gruppe, 4 Patienten (0,7 %) in der 25-mgBrensocatib-Gruppe und 7 Patien-
ten (1,2 %) in der Placebogruppe auf [17].
Non-CF-Bronchiektasen sind gekennzeichnet durch eine dauerhafte Erweiterung der Bronchien und Bronchiolen sowie einen Teufelskreis aus chronischer Infektion und Entzündung, Sekretverhalt und Schädigung der Atemwege, der letztlich zum Tod führt, denn bislang war die Erkrankung nicht heilbar. Hoffnung machen die Ergebnisse der ASPEN-Studie, die die antientzündliche Wirkung von Brensocatib (Brinsupri®) untersuchte, dem ersten Wirkstoff, der gezielt in den der BronchiektasenErkrankung zugrundeliegenden entzündlichen Prozess eingreift und damit die Erkrankung ursächlich behandeln kann. Wie die Ergebnisse zeigen, kann dieser DPP-1-Inhibitor die Verschlechterung der Lungenfunktion deutlich verlangsamen und damit auch die Lebensqualität der Betroffenen verbessern.
Literatur
1 Martinez-Garcia MA et al. Kompass Pneumol 2023;11:44-50
2 Insmed Inc. Data on File. Claims data analysis (InGEF database) on non-cystic fibrosis bronchiectasis in Germany: Final
Report. IGES Institute Berlin – September 2024
3 Ringshausen FC et al. Pneumologie 2024;78:833-899
4 Nigro M et al. Eur Respir Rev 2024; 33:240091
5 Chalmers JD et al. ERJ Open Res 2016;2:00081-2015
6 Chalmers JD et al. Am J RespirCritCare Med 2018;197:1410-1420
7 Deutsche Gesellschaft für Pneumologie und Beatmungsmedizin e. V. (DGP), Management erwachsener Patientinnen und Patienten mit Bronchiektasen-Erkrankung, Version 1.2, Mai 2024. www.awmf. org/service/awmf-aktuell/managementerwachsener-patienten-mit-bronchiektasen-erkrankung
8 Flume PA et al. Lancet 2018:392:880-890
9 Smith MP. Can Med Assoc J 2017;189: E828–E835
10 Brill SE et al. RespirRes 2015;16
11 De Angelis A et al. Eur Respir Rev 2024; 33:240085
12 Chalmers JD et al. Nat Rev Dis Primers 2018;4:45
13 Olveira C et al. Qual Life Res 2013;22: 597-605
14 Polverino E et al. Eur Respir J 2017;50: 1700629
15 Fachinformation Brinsupri®; Stand: November 2025
16 European Medicines Agency (EMA). First treatment for serious chronic lung disease, Oktober 2025. https://www.ema. europa.eu/en/news/first-treatment-serious-chronic-lung-disease
17 Chalmers JD et al. NEJM 2025;392: 1569-1581
18 Chalmers JD et al. NEJM 2020;383: 2127-2137
Anschrift der Verfasserin: Brigitte Söllner Medizinjournalistin und Wissenschaftliche Lektorin Lärchenweg 10 91058 Erlangen brigitte.soellner@online.de
Die eosinophile Ösophagitis (EoE) ist eine chronische, lokale immunvermittelte Erkrankung des Ösophagus. Kennzeichnend sind eine ösophagiale Dysfunktion sowie Eosinophilie. Zusammen mit der gastroösophagealen Refluxkrankheit (GERD) gehört die EoE zu den häufigsten Erkrankungen der Speiseröhre. Beide Entitäten können unabhängig voneinander koexistieren oder sich bidirektional beeinflussen. Unbehandelt verläuft die EoE progressiv und führt in der Regel zu einem Umbau des Ösophagus mit Strikturen und Funktionsstörungen [1]. Um das zu verhindern, sind eine frühzeitige Diagnose und Therapieeinleitung notwendig.
Adaptationsstrategien der Betroffenen können die Symptome maskieren
Eine Herausforderung für die Diagnose sind das oft angepasste Essverhalten und andere Adaptationsstrategien [1]. Daher sollten bei der klinischen Evaluation gezielte Fragen dazu gestellt werden (Tab. 1) [2].
Die häufigsten Symptome bei Jugendlichen und Erwachsenen sind Dysphagie und Bolusobstruktionen [1]. Im Fall von Bolusobstruktionen ist daher zusätzlich
IMPAKTS-Fragen zum Essverhalten und anderen Adaptationsstrategien:
• Immer der / die Letzte am Tisch?
• Meiden von fester Nahrung?
• Pürieren von Mahlzeiten, Kleinschneiden?
• Aversion / Abneigung gegen Tabletten?
• Kauen exzessiv und Einspeicheln?
• Trinkmengen erhöht bzw. „Nachtrinken“?
• Sozialer Rückzug, Kommunikation und Essen sind eine Herausforderung?
Tabelle 1: Adaptationsstrategien der Patienten (adaptiert nach Hoerning A et al. [2]).
zur endoskopischen Entfernung auch eine histologische Evaluation unablässig, um eine eventuell zugrundeliegende Erkrankung diagnostizieren zu können [1, 3]. Wie eine dänische Kohortenstudie zeigte, verlassen jedoch die meisten Betroffenen das Krankenhaus ohne Diagnose. Bei diesen Patienten ist das Risiko hoch, dass eine EoE übersehen wurde [4].
Topische Kortikosteroide als erste Wahl für die Induktions- und Erhaltungstherapie
Eine aktive EoE sollte immer auch dauerhaft behandelt werden. The-
rapieziele sind die Induktion einer klinischen und histologischen Remission sowie der langfristige Remissionserhalt und damit einhergehend die Vermeidung von Komplikationen wie Bolusobstruktionen und Strikuren sowie eine Verbesserung bzw. Normalisierung der Lebensqualität [1]. In den Empfehlungen der Deutschen Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten (DGVS) stehen topische Steroide wie die orodispersible Budesonid-Schmelztablette Jorveza® an erster Stelle bei der Behandlung erwachsener EoE-Patienten – und zwar sowohl in der Induktions- als auch in der Erhaltungstherapie. Protonenpumpenhemmer (PPI) und Eliminationsdiäten erhielten lediglich eine „Kann“-Empfehlung [1].
Wenn eine konventionelle Therapie versagt oder nicht in Betracht kommt, steht auch ein Biologikum zur Verfügung. Da die EoE nach Absetzen der antiinflammatorischen Therapie rezidiviert, sollte generell eine remissionserhaltende Therapie erfolgen [1].
Hohe und stabile Remissionsraten unter orodispersiblem Budesonid
Nach einer sechs- bis zwölfwöchigen Induktionstherapie mit
retrospektive Kohortenstudie aus zwei deutschen
bestätigt die in den klinischen Studien gezeigte Wirksamkeit und Sicherheit von Jorveza® in der Induktions- und Erhaltungstherapie über bis zu 104 Wochen im Real -World-Setting (Abb) 3
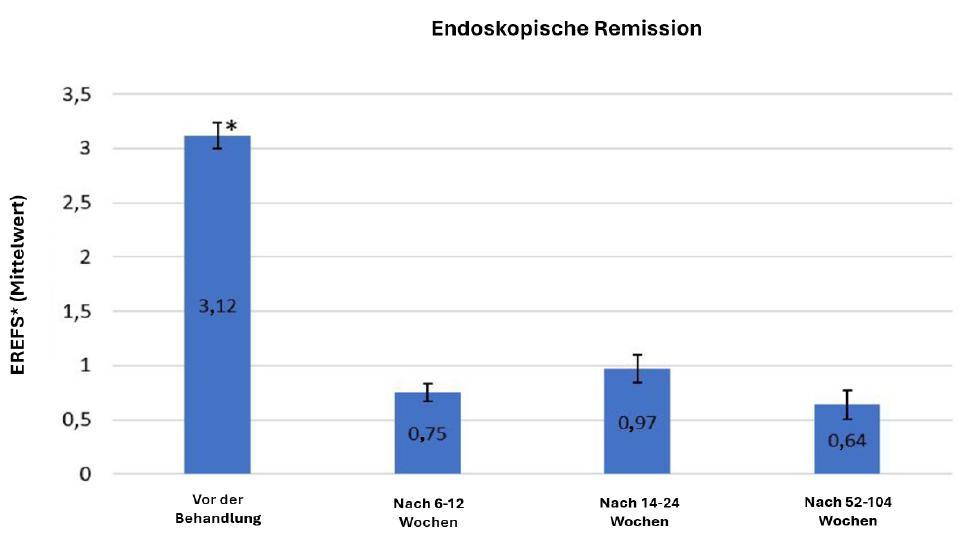
Abbildung 1: Endoskopische Remission unter der Therapie mit orodispersiblem Budesonid (Jorveza®). * EREFS: Akronym für Exsudate, Ringe, Ödeme, Furchen und Strikturen (mod. nach [8]). werden 2 Eine
Strukturierter Monitoring-Algorithmus für EoE-Management
Abb.: Endoskopische Remission. Modifiziert nach Weisfeld M et al. Adv Gastroenterol 20253
*EREFS: Akronym Exsudate, Ringe, Furchen, Strikturen
Jorveza® zeigten sich hohe Raten einer klinisch-histologischen Remission [5]. Die Remissionsraten konnten auch in der Erhaltung über 48 Wochen stabil gehalten werden [6]. Wie aktuelle Langzeiterfahrungen für die Erhaltungstherapie aus einer offenen Verlängerungsstudie über bis zu 3 Jahren belegen, konnte die klinisch-histologische Remission bei der Mehrheit der Patienten mit orodispersiblem Budesonid bis zu 96 Wochen und bei Patienten mit ununterbrochener Therapie bis zu 144 Wochen aufrechterhalten werden [7]. Eine vor Kurzem publizierte retrospektive Kohortenstudie aus zwei deutschen Zentren bestätigt die in den klinischen Studien gezeigte Wirksamkeit und Sicherheit von Jorveza® in der Induktions- und Erhaltungstherapie über bis zu
104 Wochen im Real-World-Setting (Abb. 1) [8].
Strukturierter MonitoringAlgorithmus für das EoEManagement
Ein internationales Gremium von EoE-Experten hat diesen strukturierten Monitoring-Algorithmus bestätigt [9].
Fabian Sandner, Nürnberg
Die DGVS-Leitlinien empfehlen, die Induktionstherapie nach 8 bis 12 Wochen klinisch, endoskopisch und histologisch zu reevaluieren. Bei klinisch-histologischer Remission sollte die Erhaltungstherapie mit dem in der Induktionstherapie erfolgreichen Behandlungsprinzip eingeleitet werden. Wenn keine Remission erreicht wurde, wird ein Therapiewechsel oder im Fall von symptomatischen Strikturen eine Dilatation/Bougierung empfohlen . Die Effektivität der anschließenden Erhaltungstherapie sollte alle ein bis zwei Jahre reevaluiert werden.1 Ein internationales Gremium von EoE-Expert*innen hat diesen strukturierten Monitoring-Algorithmus bestätigt.4
Die DGVS-Leitlinien empfehlen, die Induktionstherapie nach 8 – 12 Wochen klinisch, endoskopisch und histologisch zu reevaluieren. Bei klinisch-histologischer Remission sollte die Erhaltungstherapie mit dem in der Induktionstherapie erfolgreichen Behandlungsprinzip eingeleitet werden. Wenn keine Remission erreicht wurde, wird ein Therapiewechsel oder im Fall von symptomatischen Strikturen eine Dilatation/Bougierung empfohlen. Die Effektivität der anschließenden Erhaltungstherapie sollte alle 1 – 2 Jahre reevaluiert werden [1].
Literatur
1 Madisch A et al. Z Gastroenterol 2023; 61:862-933
2 Hoerning A et al. Dtsch Arztebl Int 2025; 122:195-202
3 Birk M et al. Endoscopy 2016; 48:489496
4 Terkelsen JH et al. BMC Gastroenterol 2024;24:3
5 Lucendo AJ et al. Gastroenterology 2019; 157:74-86
6 Straumann A et al. Gastroenterology 2020;159:1672-1685
7 Biedermann L et al. Clin Gastroenterol Hepatol 2025;23:2144-2154
8 Weisfeld M et al. Ther Adv Gastroenterol 2025;18:1-12
9 von Arnim U et al. Clin Gastroenterol Hepatol 2023; 21:2526-2533
Wirkfluktuationen unter Levodopa, die mit Fortschreiten der ParkinsonKrankheit bei fast allen Patienten auftreten, sind eine Herausforderung im Therapiemanagement und eine große Belastung für die Betroffenen und ihre Angehörigen. Insbesondere OFF-Episoden beeinträchtigen die Lebensqualität oft erheblich. Eine Add-on-Therapie mit Opicapon (Ongentys®) kann die ON-Zeit effektiv verlängern und Wearing-OFFs hinauszögern. Studienergebnissen zufolge profitieren Patienten mit Wirkfluktuationen bereits von Ongentys® , wenn sie erst dreimal täglich Levodopa einnehmen. Durch die Anwendung des schnell wirkenden Apomorphin-Sublingualfilms Kynmobi® kann zudem die Zeit bis zum ON gezielt verkürzt werden – dies schließt eine bislang häufig vorhandene therapeutische Lücke.
OFF-Episoden immer ernst nehmen
Levodopa (L-Dopa) ist der Goldstandard in der Parkinson-Therapie und wird von nahezu allen Patienten benötigt [1]. Um den peripheren Abbau des Wirkstoffs zu reduzieren, wird er mit einem Dopa-Decarboxylase-Inhibitor (DDCI) wie Carbidopa kombiniert [2]. Doch nach häufig zunächst gutem Ansprechen kann es mit Fortschreiten der Erkrankung zu zunehmenden Schwankungen der Levodopa-Plasmaspiegel kommen, welche die Wirksamkeit beeinträchtigen.
Nach 2,5 Jahren Erkrankung leiden bereits mehr als 40 % der Parkinson-Patienten an Wearing-OFF-Zuständen und damit verbundenen motorischen Fluktuationen [3]. Derartige Wirkfluk-
tuationen erschweren das Therapiemanagement und schränken die Lebensqualität der Betroffenen teils erheblich ein [4]. Vor allem bei fortgeschrittener Erkrankung sind plötzliche Wirkfluktuationen möglich, die unerwartet innerhalb kurzer Zeit zu einer OFF-Episode führen und für Patienten und deren Angehörige sehr belastend sein können [5, 6]. Etwa zwei Drittel der Parkinson-Patienten befinden sich täglich 2 oder sogar deutlich mehr Stunden in OFF-Zuständen [7]. Diese umfassen bereits den Beginn eines Wearing-OFFs und nicht nur die Zeit bis zum ON (Time-to-ON) [8].
Mit Ongentys® früh die ON-Zeit verlängern
Ein sinnvoller therapeutischer Ansatz für die Reduktion motorischer Fluktuationen ist die Hemmung der Catechol-O-Methyltransferase (COMT) mit einem Inhibitor wie Ongentys® [9]. Die Rationale hierbei: Trotz Kombination mit einem DDCI werden immer noch etwa 90 % einer Levodopa-Dosis über die COMT abgebaut und können daher nicht therapeutisch genutzt werden [10]. Ongentys® kann durch seinen Wirkmechanismus das bereits im System vorhandene L-Dopa besser nutzbar machen,
dessen Bioverfügbarkeit erhöhen und die L-Dopa-Plasmaspiegel glätten [11]. Dadurch lassen sich Wearing-OFFs reduzieren bzw. hinauszögern und die ON-Zeit verlängern. Dabei kann es sich lohnen, bereits früh an Ongentys® zu denken, da der frühe Einsatz dieses COMT-Hemmers auch bei Patienten mit motoriscshen Fluktuationen von weniger als 2 Jahren und mit erst 3 täglichen L-DopaEinnahmen sinnvoll ist. Dies zeigen die Ergebnisse des ADOPTION-Studienprogramms, wonach bei solchen frühen Patienten die einmal tägliche Add-on-Gabe von Ongentys® der zusätzlichen Gabe von 100 mg Levodopa/DDCI deutlich überlegen ist [12]. Laut den gepoolten Daten der beiden PhaseIV-Studien reduzierte Ongentys® die OFF-Zeit nach 4 Wochen signifikant um täglich 29,0 Minuten stärker als die Extragabe von LDopa (–62,8 vs. –33,8 Minuten; p = 0,022). Damit einhergehend verlängerte sich die ON-Zeit durch Ongentys® um zusätzliche 20,4 Minuten am Tag (64,2 vs. 43,8 Minuten; p = 0,17) [13].
In den Studien schnitt Ongentys® auch bei der Beurteilung durch die Patienten und die behandelnden Ärzte (gemessen anhand des PGI-I und CGI-I) besser ab als die zusätzliche Gabe von L-Dopa [13]. Außerdem zeichnet sich Ongen-
tys® laut der aktuellen S2k-Leitlinie „Parkinson-Krankheit“ der DGN dadurch aus, der am besten verträgliche COMT-Hemmer zu sein [2]. Allerdings wird das Potenzial einer nichtinvasiven Therapie wie Ongentys® immer noch unzureichend ausgeschöpft.
Kynmobi®: Zeit bis zum ON gezielt verkürzen und therapeutische Lücke schließen
Bei den schnellwirkenden Therapien, die ergänzend zur Parkinson-Basistherapie zum Einsatz kommen können, um eine akute OFF-Episode zügig zu durchbrechen, hat es einen Paradigmenwechsel gegeben: Während sie lange Jahre bei Auftreten von Wirkfluktuationen erst nach der Optimierung der Basistherapie zum Einsatz kamen, werden schnellwirkende Therapien heute immer öfter parallel in Erwägung gezogen [14, 15], um den Anteil täglicher OFF-Episoden möglichst effektiv zu reduzieren. Das ist vor allem bei morgendlichen OFF-Episoden der Fall, wenn die erste L-Dopa-Dosis erst verzögert wirkt (sog. Delayed-ONs) oder bei Dose Failures, bei denen L-Dopa gar nicht wirkt. In diesen Fällen sollten schnellwirkende Therapien, die diese OFF-Episoden innerhalb einer kurzen Zeit überwinden können, spätestens nach 15 Minuten einen Wirkeffekt haben. Dabei sollte wenn möglich der Gastrointestinaltrakt umgangen werden, da Bewegungsstörungen im gesamten Magen-Darm-Trakt wie eine oropharyngeale Dysphagie, ösophageale Motilitätsstörung oder Gastroparese die L-Dopa-Resorption behindern und somit zu Wirkfluktuationen führen können [16]. Eine
in dieser Situation empfehlenswerte schnellwirkende Therapie ist der Sublingualfilm Kynmobi®, bei dem der Wirkstoff Apomorphin über die Mundschleimhaut aufgenommen und so der Gastrointestinaltrakt und First-Pass-Stoffwechsel umgangen wird. Kynmobi® ist zugelassen für die intermittierende Behandlung von OFF-Episoden bei erwachsenen Patienten mit Parkinson-Krankheit, die durch oral angewendete Antiparkinsonmittel nicht ausreichend eingestellt sind [17].
In der Zulassungsstudie verbesserte Kynmobi® im Vergleich zu Placebo bei Anwendung zu Beginn einer OFF-Episode bereits nach 15 Minuten die Motorik – gemessen mittels MDS-UPDRS Part III – signifikant (–6,4 vs. –3,0; p = 0,039;) [18]. So konnten innerhalb von 30 Minuten etwa 80 % der OFF-Episoden beendet und in einen vollständigen ON-Zustand überführt werden, wobei die Wirkung bis zu 90 Minuten anhielt [18]. Auch in der Langzeitanwendung bestätigten sich die Effektivität und die konstante Wirksamkeit: Über 48 Wochen führte Kynmobi® mehr als Dreiviertel der OFF-Episoden in einen vollständigen ON-Zustand über [19, 20]. Damit schließt Kynmobi® die therapeutische Lücke und verkürzt gezielt die Zeit bis zum ON, unabhängig von gastrointestinalen Einschränkungen, der Art der sonstigen Parkinson-Therapie und der Ursache der OFFEpisoden [17, 18]. Von Vorteil dabei ist die einfache Anwendung des Sublingualfilms, die der Patient bis zu fünfmal täglich selbst durchführen kann [17].
Generell sollten die verfügbaren schnellwirksamen Therapien bereits beim Auftreten der ersten Wirkfluktuationen mit dem Patienten besprochen, ihre Wirksamkeit
individuell getestet und ihre Anwendung im Medikamentenplan festgelegt werden, damit die Patienten nicht unnötig lange in OFFEpisoden verbringen müssen.
Elisabeth Wilhelmi, München
Literatur
1 Jenner P et al. Expert Review of Neurotherapeutics 2021; 21: 1019-1033
2 https://register.awmf.org/de/leitlinien/detail/030-010
3 Stocchi F et al. Parkinsonism Relat Disord 2014;20:204-211
4 Politis M et al. Mov Disord 2010;25: 1646-1651
5 Chou KL et al. Parkinsonism Relat Disord 2018;51:9-16
6 Damier P et al. Parkinsons Dis 2022; doi: 10.1155/2022/1800567
7 Michael J. Fox Foundation for Parkinson’s Research. Capturing and elevating the patient voice (2014)
8 Merims D et al. Clin Neuropharmacol 2003;26:196-198
9 Fachinformation Ongentys®; Stand: März 2022
10 Rocha JF et al. Eur J Clin Pharmacol 2014;70:1059-1071
11 Ferreira JJ et al. Mov Disord 2022;37: 2272-2283
12 Lee JY et al. Mov Disord Clin Pract 2024;11: 655-665
13 Ferreira JJ et al. J Neurol 2024;271: 6729-6738
14 Isaacson SH et al. Clin Park Relat Disord 2022;7:100161
15 Pahwa R et al. Neurol Ther 2023;12: 1033-1049
16 Leta V et al. Eur J Neurol 2023;30:14651480
17 Fachinformation Kynmobi®; Stand: Oktober 2023
18 Olanow CW et al. Lancet Neurol 2020; 9:135-144
19 Kassubek J et al. J Neurol 2024;271: 3554-3570
20 Factor SA et al. Mov Disord 2021; 36(Suppl 1): Poster 384
Das Multiple Myelom (MM) ist eine aggressive und derzeit nicht heilbare Form von Blutkrebs, die durch entartete Plasmazellen im Knochenmark gekennzeichnet ist [1]. In Deutschland erhalten jedes Jahr etwa 7.000 bis 8.000 Menschen diese Diagnose, in Europa sind es jährlich über 50.000 neue Fälle und weltweit rund 188.000 [2, 3, 4].
5 Jahre nach der Diagnose ist nur noch etwa die Hälfte der von MM betroffenen Patienten am Leben und die meisten von ihnen erhalten aufgrund von Rezidiven 4 oder mehr Therapielinien [5].
Die Krankheitsverläufe unterscheiden sich zwar individuell, jedoch sind Rezidive fast unvermeidbar. Daten aus der Versorgungsforschung zeigen, dass Patienten mit einem rezidivierten und refraktären Multiplen Myelom (RRMM) häufig bereits nach wenigen Therapielinien refraktär gegenüber einer Kombination der 3 Haupttherapieklassen (Proteasom-Inhibitoren, immunmodulatorische Wirkstoffe und Anti-CD38-Antikörper) sind [6].
Eine neuere Behandlungsmöglichkeit in dieser Situation ist Elranatamab (Elrexfio®), das im Dezember 2023von der Europäischen Kommission die bedingte Zulas-
Elranatamab (Elrexfio®) ist eine subkutan zu verabreichende, gegen das B-Zell-Reifungsantigen (BCMA) und CD3 gerichtete, bispezifische Antikörper (BsAb)-Immuntherapie, die an BCMA auf den Myelomzellen und an CD3 auf den T-Zellen bindet, wodurch die T-Zellen aktiviert werden und die Myelomzellen abtöten [7].
ermöglicht verlängerte Therapiepausen ohne Wirksamkeitsverlust
sung für die Behandlung von Erwachsenen mit RRMM bekam, die zuvor bereits mindestens 3 Therapien erhalten haben, darunter einen immunmodulatorischen Wirkstoff, einen Proteasom-Inhibitor und einen Anti-CD38-Antikörper, und die während der letzten Therapie eine Krankheitsprogression gezeigt haben [7].
Langanhaltende Remission trotz Therapieunterbrechung
Auf dem ASH-Kongress im Dezember 2025 wurde eine retrospektive Analyse der Phase-IIStudie MagnetisMM-3-Studie vorgestellt, die zeigt, dass bei Patienten mit RRMM verlängerte Behandlungspausen möglich sind, ohne die Wirksamkeit der Therapie mit Elranatamab zu gefährden [8]. Untersucht wurde eine Subgruppe von 28 Patienten, die eine verlängerte Therapiepause von mehr als 6 aufeinanderfolgenden Monaten hatten oder die Therapie abbrachen, aber weiterhin hinsichtlich der Wirksamkeit beobachtet wurden. Die mediane Behandlungsdauer vor der Unterbrechung bzw. dem Absetzen betrug 12,1 Monate, die mediane Dauer der behandlungsfreien Phase lag bei 15,5 Monaten [8].
Die Langzeitdaten zeigen: Das mediane progressionsfreie Überleben wurde nicht erreicht (95%-KI: 38,5 bis nicht abschätzbar [NE]), die Wahrscheinlichkeit, nach 36 Monaten ereignisfrei zu sein, betrug 79,4 % (95%-KI: 57,0–90,9). Auch das mediane Gesamtüberleben wurde nicht erreicht (95%-KI: NE–NE), die Überlebenswahrscheinlichkeit nach 36 Monaten betrug 84,2 % (95%-KI: 63,0–93,8) [8].
Von den 28 untersuchten Patienten erlitten lediglich drei eine Krankheitsprogression – diese trat 1 – 3 Jahre nach der letzten Dosis auf. Zwei Patienten setzten die Elranatamab-Therapie nach Unterbrechungen von circa 12 bzw. 14 Monaten erfolgreich fort. Beide hatten zu Beginn der längeren Unterbrechung eine komplette Remission (≥CR) erreicht und befanden sich zum Datenschnitt weiterhin in ≥CR.
Zusammenfassend zeigen die Daten, dass die Mehrheit der Patienten trotz verlängerter Behandlungsunterbrechung ein anhaltendes klinisches Ansprechen aufwies. Dies unterstreicht die Dauerhaftigkeit der durch Elranatamab induzierten Wirkung und unterstützt die Durchführbarkeit gezielter Therapieunterbrechungen, beispielsweise zur Handhabung von Nebenwirkungen [8].
Real-World-Daten bestätigen Wirksamkeit in der Versorgungsrealität
Auf dem ASH wurden außerdem die Ergebnisse einer internationale Real-World-Analyse vorgestellt, die 79 Patienten mit RRMM aus 7 Ländern untersuchte, die außerhalb klinischer Studien mit Elranatamab behandelt wurden [9]. Die im Median 67 Jahre alten Studienteilnehmer hatten eine umfassende Vorbehandlung mit im Median 5 Therapielinien erhalten, 35 % davon auch eine gegen das B-Zell-Reifungsantigen (BCMA)-gerichtete Behandlung. Die Gesamtansprechrate auf die Elranatamab-Therapie betrug 53 % in der gesamten Kohorte und 67 % bei den Patienten ohne vorherige BCMA-gerichtete Therapie (BCMAnaiv). Nach 12 Monaten erreichten 67 % der BCMA-naiven Patienten ein progressionsfreies Überleben und die Gesamtüberlebensrate betrug 70 %. Diese Ergebnisse entsprechen weitgehend den Daten der MagnetisMM-3-Studie, obwohl fast die Hälfte der Patienten (48 %) die Einschlusskriterien der Zulassungsstudie nicht erfüllt hätte [9]. Die Analyse identifizierte auch prognostische Faktoren für schlechtere Behandlungsergebnisse: Eine vorherige Behandlung mit einer gegen BCMA gerichteten Therapie, niedrige Thrombozytenwerte, ein hohes zytogenetisches Risiko und eine Penta-Refraktärität waren mit geringeren Ansprechraten und kürzerem OS assoziiert [9].
Konsistentes Sicherheitsprofil
In beiden Analysen zeigte Elranatamab ein konsistentes Sicherheitsprofil, das den bereits zuvor beobachteten Daten entsprach. In der Real-World-Analyse erlitten
41 % der Patienten ein Zytokinfreisetzungssyndrom (CRS), überwiegend in geringgradiger Ausprägung (Grad 1: 35 %, Grad 2: 5 %), wobei keine schweren Fälle (≥Grad 3) beobachtet wurden. Die meisten CRS-Ereignisse traten bei der ersten Step-up-Dosis auf. Ein Immuneffektor-assoziiertes Neurotoxizitätssyndrom (ICANS) wurde in 5 % der Fälle beobachtet [9]. Insgesamt erwies sich das Sicherheitsprofil von Elranatamab als planbar und gut kontrollierbar [7, 10].
Das klinische Forschungsprogramm MagnetisMM untersucht den Einsatz von Elranatamab bei Patienten mit Multiplem Myelom (MM), vom RRMM bis hin zum neu diagnostizierten MM. MagnetisMM-3 (NCT04649359) ist eine offene, multizentrische, nicht randomisierte Phase-II-Studie zur Elranatamab-Monotherapie bei Patienten mit Multiplem Myelom, die gegenüber mindestens einem Proteasom-Inhibitor, einem immunmodulatorischen Wirkstoff und einem Anti-CD38-Antikörper refraktär sind. Die Studie wurde mit 2 Kohorten ausgerollt: eine mit und eine ohne vorherige Behandlung mit einem gegen das B-ZellReifungsantigen gerichteten Antikörper-Wirkstoff-Konjugat oder chimären Antigenrezeptor-T-Zelltherapie. Die Teilnehmer erhielten Elranatamab subkutan in Form von 2 initialen Step-up-Dosierungen, gefolgt von einer wöchentlichen 76 mg-Injektion. Primärer Endpunkt ist die objektive Ansprechrate nach verblindeter unabhängiger zentraler Bewertung. Zu den wichtigsten sekundären Endpunkten gehören die Dauer des Ansprechens, das progressionsfreie Überleben,
die Rate der minimalen Resterkrankung, das Gesamtüberleben und die Sicherheit.
Weitere laufende zulassungsrelevante Studien vergleichen Elranatamab mit bisherigen Therapiestandards – sowohl als Monotherapie als auch in Kombination mit Standardoder neuartigen Therapien: MagnetisMM-5 im exponierten Setting mit 2 Arzneimittelklassen (ClinicalTrials.gov. NCT05020236), MagnetisMM-6 bei Patienten mit neu diagnostiziertem MM, bei denen eine Stammzelltransplantation nicht infrage kommt (ClinicalTrials.gov. NCT05623020), MagnetisMM-7 bei neu diagnostizierten MM-Patienten nach der Transplantation (ClinicalTrials.gov. NCT05317416. O) und MagnetisMM-32 bei Patienten mit vorheriger Anti-CD38-gerichteter Therapie (ClinicalTrials.gov. NCT06152575).
Brigitte Söllner, Erlangen
Literatur
1 Multiple Myeloma Research Foundation (MMRF). What is Multiple Myeloma? https://themmrf.org/multiple-myeloma
2 Myeloma Patients Europe. Myeloma – A Patients’ Guide. www.mpeurope.org/wpcontent/uploads/2023/01/Myeloma-Patients-Guide.pdf
3 Deutsches Krebsforschungszentrum DKFZ. Multiples Myelom (Knochenmarkkrebs). www.krebsinformationsdienst.de/tumorarten/mulitples-myelom/ index.php
4 World Health Organization. Globocan 2022: Multiple Myeloma. https://gco.iarc. who.int/media/globocan/factsheets/ cancers/35-multiple-myeloma-fact-sheet. pdf
5 Dimopoulos MA et al. Clin Lymphoma Myeloma Leuk 2022;22:460-473
6 Guillaume X et al. HemaSphere 2023;7: e8647215
7 Fachinformation Elrexfio®; Stand: November 2025
8 Lesokhin et al. Abstract presented at ASH 2025; abs25-8338
9 Popat et al. Abstract presented at ASH 2025; abs25-11599
10 Lesokhin AM et al. Nat Med 2023;29: 2259-2267
Im November 2025 hat die Europäische Kommission Elinzanetant unter dem Markennamen Lynkuet® die Marktzulassung in der Europäischen Union erteilt. Elinzanetant ist eine duale auf Neurokinin-Rezeptoren gerichtete Therapie. Der NK-1- und NK3-Rezeptor-Antagonist beeinflusst das Temperaturregulierungszentrum im Gehirn und ermöglicht damit eine hormonfreie Behandlung von moderaten bis schweren vasomotorischen Symptomen (VMS), die in den Wechseljahren als Hitzewallungen und Nachtschweiß auftreten oder durch eine adjuvante endokrine Therapie (AET) im Zusammenhang mit Brustkrebs verursacht werden [1]. Die europäische Zulassung für diese beiden Indikationen basiert auf den positiven Ergebnissen des klinischen Phase-III-Programms OASIS (OASIS-1–4), in dem Elinzanetant bei Frauen in den Wechseljahren sowie bei Patientinnen, die eine endokrine Therapie wegen Brustkrebs erhielten, signifikant die Häufigkeit und Schwere von moderaten bis schweren VMS reduzierte und dabei ein positives Sicherheitsprofil zeigte [2, 3, 4].
Hitzewallungen und Nachtschweiß plagen viele Frauen in den Wechseljahren
Menopausale Symptome wie Hitzewallungen, Nachtschweiß und Schlafstörungen können die Lebensqualität von Frauen erheblich beeinträchtigen. Nach Schätzungen werden bis 2030 weltweit 1,2 Milliarden Frauen in den Wechseljahren sein [5]. Bis zu 80 % der Frauen haben während dieser menopausalen Transition Beschwerden [6]. In Europa berichten etwa 40 % dieser Frauen von moderaten
bis schweren Hitzewallungen, die ihr tägliches Leben, ihr Wohlbefinden und ihre Leistungsfähigkeit erheblich belasten [7].
Menopausale Symptome als Nebenwirkungen der endokrinen Brustkrebstherapie
VMS, insbesondere Hitzewallungen, können auch durch eine adjuvante endokrine Therapie zur Behandlung oder Prävention von Brustkrebs verursacht werden. Etwa 70 % der Mammakarzinome werden als hormonrezeptorpositiv (HR+) eingestuft [8, 9] und mit einer entsprechenden endokrinen Therapie behandelt. Diese kann jedoch auch VMS induzieren [10, 11], die die Lebensqualität, aber auch die Therapietreue beeinträchtigen, schlimmstenfalls so sehr, dass die Patientinnen die für sie wichtige Behandlung abbrechen.
Mögliche Risiken der Langzeit-Hormontherapie
Zur Linderung der menopausalen Symptome werden häufig verschiedene Präparate mit Östrogen und Gestagen verordnet, die die in den Wechseljahren fehlenden weiblichen Geschlechtshormone ersetzen sollen. Diese Hormonersatztherapie sollte aber nur kurzfristig angewendet werden, da sie bei längerer – vor allem oraler –
Anwendung das Risiko für Brustkrebs, venöse Thromboembolien, Schlaganfall und Gallenblasenerkrankungen, die einen operativen Eingriff erfordern, erhöhen [12]. Nutzen und Risiken einer Hormonersatztherapie sollten daher immer sorgfältig abgewogen werden.
Gezielte hormonfreie Therapie reduziert Anzahl und Intensität von VMS signifikant
Den bislang ungedeckten Bedarf an einer langfristigen, sicheren, wirksamen und hormonfreien Therapie der vasomotorischen Symptome, unter denen Frauen in den Wechseljahren leiden, kann der Neurokinin-Rezeptor-Antagonist Elinzanetant (Lynkuet®) decken, der seine Wirksamkeit und Sicherheit in den Studien OASIS-1, -2, -3 und -4 unter Beweis stellte [2, 3, 4].
Wirksamkeit bei VMS in der Menopause
Die randomisierten, doppelblinden, placebokontrollierten Phase-III-Studien OASIS 1, 2 und 3 untersuchten die Wirksamkeit und Sicherheit von Elinzanetant bei Frauen mit moderaten bis schweren vasomotorischen Symptomen in den Wechseljahren [2, 3].
In OASIS-1 und -2 wurden 396 bzw. 400 postmenopausale Frauen
Elinzanetant (Lynkuet®) ist ein nicht hormonell wirksamer, selektiver Neurokinin-1 (NK-1)- und -3 (NK-3)-Rezeptor-Antagonist. Er blockiert die Signalübertragung an hypothalamische Neuronen, die sowohl NK-1- als auch NK-3-Rezeptoren und deren Liganden exprimieren. Diese als Kisspeptin/NeurokininB/Dynorphin (KNDy) bezeichneten Neuronen spielen eine zentrale Regulation bei der Thermo- und Schlafregulierung. Sinkende Östrogenspiegel aufgrund der natürlichen Wechseljahre oder einer endokrinen Therapie führen zu einer Hyperaktivität der KNDy-Neuronen und zu einer Dysregulation des thermoregulatorischen Zentrums, wodurch vasomotorische Symptome (VMS) ausgelöst werden. Indem Elinzanetant verhindert, dass Neurokinin B an seine Rezeptoren bindet, wird die Überaktivität der KNDy-Neuronen gedämpft und die Häufigkeit bzw. Schwere der VMS (v.a. Hitzewallungen, Nachtschweiß) reduziert.
Lynkuet® liegt als 60-mg-Weichkapsel vor. Die empfohlene Tagesdosis beträgt 120 mg (zwei 60-mg-Kapseln) Elinzanetant, eingenommen vor dem Schlafengehen [1].
zwischen 40 und 65 Jahren eingeschlossen, bei denen mindestens 50 moderate bis schwere VMS (einschließlich nächtlicher VMS) pro Woche auftraten. Die Frauen in der Elinzanetant-Gruppe erhielten einmal täglich 26 Wochen lang eine Dosis von 120 mg Elinzanetant, während die Frauen in der Kontrollgruppe zunächst 12 Wochen lang einmal täglich ein entsprechendes Placebo bekamen, gefolgt von 14 Wochen mit einer 120 mg-Dosis Elinzanetant. Nach 4 Wochen sank die durchschnittliche Anzahl mittelschwerer bis starker Hitzewallungen pro Tag bei den mit Elinzanetant behandelten Frauen in OASIS-1 gegenüber dem Ausgangswert signifikant um –7,5 und bei den Frauen, die ein Placebo erhielten, um –4,4. Nach 12 Wochen hatte sich die Anzahl der VMS unter Elinzanetant um –8,7 versus –5,5 unter Placebo verringert.
In OASIS-2 nahmen die Hitzewallungen unter Elinzanetant ebenfalls signifikant ab: nach
4 Wochen um −8,6 versus −6,1 unter Placebo und nach 12 Wochen um −10,0 versus −7,2 unter Placebo. Die Frauen in der Elinzanetant-Gruppe berichteten zudem über eine stärkere Linderung der Hitzewallungen, eine Verbesserung der Schlafstörungen und eine höhere Lebensqualität im Vergleich zu den Frauen in der Placebo-Gruppe [2]. Vergleichbare Ergebnisse erzielte die Studie OASIS-3 mit 628 postmenopausalen Frauen zwischen 40 und 65 Jahren, die über 52 Wochen randomisiert entweder Elinzanetant oder Placebo erhielten. In Woche 12 betrug die mittlere Veränderung der täglichen Häufigkeit mäßiger bis schwerer VMS gegenüber dem Ausgangswert –5,4 unter Elinzanetant und –3,5 unter Placebo. Die Ergebnisse zeigten außerdem, dass die Wirkung von Elinzanetant, d.h. die Verringerung der Häufigkeit und Schwere von menopausenbedingten Hitzewallungen, mindestens 50 Wochen anhielt [3].
Wirksamkeit bei VMS bei Hormonrezeptor-positivem Brustkrebs
Die doppelblinde, randomisierte, placebokontrollierte Studie OASIS-4 untersuchte die Wirksamkeit und Sicherheit von Elinzanetant zur Behandlung vasomotorischer Symptome bei 474 Patientinnen im Alter von 18 – 70 Jahren mit Hormonrezeptor-positivem Brustkrebs, die eine endokrine Therapie erhielten oder aufgrund eines hohen Brustkrebsrisikos präventiv mit Hormonen behandelt wurden. Die Frauen wurden im Verhältnis 2:1 randomisiert und bekamen entweder einmal täglich 120 mg Elinzanetant über 52 Wochen oder einmal täglich Placebo über 12 Wochen, gefolgt von einmal täglich 120 mg Elinzanetant über 40 Wochen. Wichtigster Endpunkt war die Veränderung der mittleren täglichen Häufigkeit mittelschwerer bis schwerer VMS im Vergleich zum Ausgangswert bis Woche 4 und bis Woche 12. Die Studie erstreckte sich über 52 Wochen und enthielt eine optionale Verlängerung von 2 Jahren, die aktuell noch läuft [4].
Nach 4 Wochen Behandlung sank die durchschnittliche Anzahl täglicher mittelschwerer bis starker Hitzewallungen in der Elinzanetant-Gruppe gegenüber dem Ausgangswert um –6,5 versus –3,0 Episoden in der Placebo-Gruppe. In Woche 12 betrug die mittlere Veränderung –7,8 in der Elinzanetant-Gruppe und –4,2 Episoden unter Placebo.
Die Wirkung von Elinzanetant hinsichtlich der Reduktion der Hitzewallungen hielt mindestens 50 Wochen an. In dieser Studie berichteten Frauen, die Elinzanetant einnahmen, außerdem über eine stärkere Verbesserung ihrer Schlaf-
störungen und ihrer menopausenbedingten Lebensqualität im Vergleich zu Frauen, die ein Placebo erhielten.
In allen 4 zulassungsrelevanten klinischen Phase-III-Studien erfüllte Elinzanetant (Lynkuet®) alle primären sowie die wichtigsten sekundären Endpunkte und zeigte ein positives Sicherheitsprofil [2, 3, 4]. Die Zulassung des NK-1und NK-3-Rezeptorantagonisten erweitert zum einen die therapeutischen Optionen für Patientinnen mit belastenden wechseljahresbedingten Symptomen um eine gezielte, hormonfreie Behandlung, die die Lebensqualität und persönliche Produktivität der Frauen in dieser herausfordernden Zeit verbessern kann. Zum anderen kann der hormonfreie Wirkstoff dazu
beitragen, die menopausalen Symptome zu lindern, die als häufige Nebenwirkungen einer adjuvanten endokrinen Therapie bei Hormonrezeptor-positivem Brustkrebs auftreten und die Frauen oft zu einem Abbruch der für sie so wichtigen Behandlung zwingen.
Brigitte
Söllner, Erlangen
Literatur
1 Fachinformation Lynkuet®, aktueller Stand
2 Pinkerton JV et al. Elinzanetant for the treatment of vasomotor symptoms associated with menopause: OASIS 1 and 2 randomized clinical trials. JAMA 2024;332:1343-1354
3 Panay N et al. Elinzanetant for the treatment of vasomotor symptoms associated with menopause: a phase 3 randomized clinical trial. JAMA Intern Med 2025; 185:1319-1327
4 Cardoso F et al. Elinzanetant for vasomotor symptoms from endocrine therapy for breast cancer. N Engl J Med 2025; 393:753-763
5 Hill K. The demography of menopause. Maturitas 1996;23:113-127
6 Gold EB et al. Longitudinal analysis of the association between vasomotor symptoms and race/ethnicity across the menopausal transition: Study of Women’s Health Across the Nation. Am J Public Health 2006;96:1226-1235
7 Nappi RE et al. Global cross-sectional survey of women with vasomotor symptoms associated with menopause: prevalence and quality of life burden. Menopause 2021;28:875-882
8 World Health Organization. Breast cancer. 2024. www.who.int/news-room/factsheets/detail/breast-cancer
9 Selli C et al. Accurate prediction of response to endocrine therapy in breast cancer patients: current and future biomarkers. Breast Cancer Res 2016;18:118
10 Burstein HJ et al. Adjuvant endocrine therapy for women with hormone receptor-positive breast cancer: ASCO Clinical Practice Guideline Focused Update. J Clin Oncol 2019;37:423-438
11 Cucciniello L et al. Estrogen deprivation effects of endocrine therapy in breast cancer patients: Incidence, management and outcome. Cancer Treat Rev 2023; 120:102624
12 Bofill Rodriguez M et al. Long-term hormone therapy for perimenopausal and postmenopausal women. Cochrane Database Syst Rev 2025;11:CD004143
Inhaltsstoff-Suche und Stabilitätsrechner von Sanofi
Sanofi hat seine digitalen Angebote für Fachkreisangehörige und Patienten auf www.sanofimedicalinformation.com um eine Inhaltsstoffsuche erweitert. Mit diesem Tool können Apotheker, PTAs, medizinisches Fachpersonal und Patienten gezielt prüfen, ob Arzneimittel oder Impfstoffe von Sanofi Allergene oder andere relevante Inhaltsstoffe enthalten. Häufig gesuchte Begriffe in der Inhaltsstoffsuche sind zum Beispiel Latex, tierische Bestandteile, Gluten oder Laktose. Die Informationen sind frei zugänglich und stehen rund um die Uhr zur Verfügung, was besonders bei dringenden Fragen

QR-Code zu sanofimedicalinformation.com
außerhalb der üblichen Geschäftszeiten wie z.B. während des Apothekennotdienstes hilfreich ist. Zusätzlich stehen aktuelle Fach- und Gebrauchsinformationen, Anwendungsvideos sowie umfassende Antworten auf häufige Fragen zu Sanofi-Produkten bereit.
Exklusiv für Fachkreisangehörige
Die deutschsprachige Plattform bietet ein weiteres praxisrelevantes Tool für Fachkreise an: einen Stabilitätsrechner. Dieses Instrument hilft bei der Entscheidung, ob ein Impfstoff nach einer Unterbrechung der Kühlkette noch verwendet werden darf – ein häufiges Szenario z.B. bei einem Kühlschrankausfall in der Apotheke oder in der Praxis. Geben Sie einfach den betroffenen Impfstoff, die Temperatur und die Dauer der Abweichung ein und der Kalkulator liefert eine wissenschaftlich fundierte Einschätzung zur weiteren Verwendbarkeit. Dies vermeidet unnötige Verwürfe und gibt Sicherheit bei der Beratung.
S. M.
Aktuelle Real-World-Daten aus Kalifornien bestätigen die Effektivität des RSVImpfstoffs Abrysvo®, der Antigene der RSV-Untergruppen A und B enthält [1], nicht nur in der ersten, sondern auch in der zweiten Saison nach der Impfung gegen das Respiratorische Synzytial-Virus: Bei geimpften über 60-Jährigen waren weniger RSV-bedingte Hospitalisierungen oder Behandlungen in der Notaufnahme wegen RSV-assoziierten Erkrankungen der unteren Atemwege erforderlich [2]. Dies ist zusammengefasst das Ergebnis einer US-amerikanischen retrospektiven Test-negativen FallKontroll-Studie, die basierend auf Daten der kalifornischen Health Maintenance Organization Kaiser Permanente die Effektivität des Präfusions-F-Impfstoffes (RSVpreF) Abrysvo®, bei Personen ab 60 Jahren untersuchte [2]. In die Studie wurden 767 Erwachsene im Alter von ≥60 Jahren eingeschlossen, die während der ersten Saison (kumulative Auswertung November 2023 bis April 2024 und November 2024 bis April 2025)* oder der zweiten Saison (November 2024 bis April 2025)** mit einer Erkrankung der unteren Atemwege hospitalisiert oder in der Notaufnahme behandelt wurden und deren respiratorischer Abstrich positiv auf RSV getestet wurde***. Die Analyse verwendete vordefinierte Kontrollgruppen, wobei
* Impfzeitpunkt: Oktober 2023 bis April 2024 und Mai 2024 bis April 2025
** Impfzeitpunkt: Oktober 2023 bis April 2024
*** Respiratorische Abstriche, die mit einem Multiplex-PCR-Test auf RSV getestet wurden (entweder mittels Standardtest oder erweiterter Probenentnahme von wiederverwendeten Abstrichen, die nicht positiv auf SARS-CoV-2 oder Influenza getestet wurden und noch nicht auf RSV getestet wurden).
der 2.
die Primäranalyse eine strenge Kontrollgruppe einbezog****. Die Ergebnisse liefern nun RealWorld-Daten zur Effektivität von Abrysvo® für die zweite RSV-Saison [2].
Anhaltende Wirksamkeit von bis zu 78 Prozent in Saison 2
Wie die Studiendaten belegen, bietet Abrysvo® auch in der zweiten Saison nach der Impfung einen anhaltend hohen Schutz sowohl vor einer RSV-assoziierten Hospitalisierung oder Behandlung in der Notaufnahme als auch vor RSVassoziierten Erkrankungen der unteren Atemwege: In der ersten Saison nach der Impfung betrug die Effektivität von Abrysvo® beim Schutz vor einer RSV-bedingten Hospitalisierung oder einer Behandlung in der Notaufnahme
**** Die strenge Kontrollgruppe (n = 1.468) umfasste Personen mit einer RSV-negativen Erkrankung der unteren Atemwege, die hospitalisiert oder in der Notaufnahme behandelt wurden und negativ auf das humane Metapneumovirus, SARS-CoV-2 und Influenza und positiv auf eine nicht-impfpräventable Ursache (z.B. Adenoviren, Rhinoviren) getestet wurden. Die breite Kontrollgruppe (n = 9.836) umfasste alle Personen mit einer RSV-negativen Erkrankung der unteren Atemwege, die hospitalisiert oder in der Notaufnahme behandelt wurden.
81 % und in der zweiten Saison nach der Impfung lag sie bei 75 %. Gegen schwere Erkrankungen der unteren Atemwege konnte Abrysvo® mit einer Effektivität von 85 % in der ersten und mit einer Effektivität von 75 % in der zweiten Saison nach der Impfung schützen. Mit Blick auf die RSV-Subtypen A und B erzielte der Impfstoff in Bezug auf RSV-bedingte Krankenhauseinweisungen oder Behandlungen in der Notaufnahme für die erste Saison jeweils eine Effektivität von 78 % für beide RSV-Subtypen. In der zweiten Saison nach der Impfung war die Effektivität mit 75 % für RSV-Subtyp A und 77 % für RSV-Subtyp B vergleichbar [2].
Real-World-Daten unterstützen die Wirksamkeitsdaten aus der Zulassungsstudie RENOIR bei Personen ab 60 Jahren
Die aktuellen Ergebnisse aus dem US-amerikanischen Versorgungsalltag zur zweiten Saison nach der Impfung mit Abrysvo® bestätigen die sehr guten Wirksamkeitsdaten der Zulassungsstudie RENOIR [3]: In dieser placebokontrollierten Phase-III-Studie mit über 36.000 Teilnehmern im Alter von ≥60 Jah-
ren schützte der Impfstoff mit einer Wirksamkeit von 88,9 % in der ersten vollständigen RSV-Saison und mit einer Wirksamkeit von 77,8 % in der zweiten vollständigen Saison gegen RSV-assoziierte Erkrankungen der unteren Atemwege mit ≥3 Symptomen. Über 2 Saisons hinweg betrug die Wirksamkeit gegen RSV-assoziierte Erkrankungen der unteren Atemwege 81,5 % [3].
STIKO empfiehlt RSVSchutzimpfung für ältere Menschen
RSV ist ein weltweit verbreiteter Erreger von akuten Erkrankungen der oberen und unteren Atemwege [4]. Das Risiko für einen schweren Verlauf einer RSV-assoziierten Erkrankung steigt mit zunehmendem Alter sowie bei bestehenden Komorbiditäten oder Immundefizienz [5]. In der vergangenen Saison 2024/25 wurden dem Robert Koch-Institut fast 70.000 RSVFälle gemeldet [6]. Zudem musste jeder dritte Erwachsene im Alter von 60 – 79 Jahren, der schwer an einer RSV-bedingten Atemwegsinfektion erkrankte, intensivmedizinisch behandelt werden [7]. Zur Reduktion der hohen Krankheitslast hat die Ständige Impfkommission (STIKO) die RSV-Schutzimpfung für ältere Menschen in ihre Empfehlungen aufgenommen und empfiehlt die RSV-Impfung für alle Personen ab einem Alter von 75 Jahren sowie für Personen in einem Alter von von 60 bis 74 Jahren, die an einer schweren Grunderkrankung leiden, die mit einem erhöhten Risiko für einen schweren Verlauf einer RSV-Erkrankung assoziiert sind, wie chronische Erkrankungen der Atmungsorgane, chronische Herz-
Kreislauf- und Nierenerkrankungen, chronische neurologische und neuromuskuläre Erkrankungen, hämatoonkologische Erkrankungen, Diabetes mellitus (mit Komplikationen) sowie schwere angeborene oder erworbene Immundefizienz [8], oder in einer Einrichtung der Pflege leben bzw. ambulante Pflegedienste sowie ambulante Betreuungsdienste in Anspruch nehmen [9].
Brigitte Söllner, Erlangen
GeriPAIN zum Schmerzmanagement bei älteren Menschen
Ab sofort steht GeriPAIN, die erste nationale und internationale S3Leitlinie zum Schmerzmanagement bei geriatrischen Patienten in allen Versorgungssettings, im AWMF-Leitlinienregister (Registernummer: 145-002) zum Download bereit. Die Entwicklung der Leitlinie wurde durch den Innovationsfonds des G-BA gefördert und von der Deutschen Gesellschaft für Geriatrie (DGG) sowie der Deutschen Schmerzgesellschaft koordiniert.
Evidenzbasierte Empfehlungen für die Schmerzerfassung und Therapie
„Die Leitlinie liefert erstmals eine strukturierte, evidenzbasierte Orientierung für die Schmerzerfassung und Therapie von Schmerzen bei älteren Menschen“, erklärte Dr. Corinna Drebenstedt, federführende Autorin der DGG.
Literatur
1 1 Fachinformation Abrysvo®; Stand: September 2025
1 Tartof SY et al. Lancet 2025; DOI: 10. 2139/ssrn.5803145
3 Walsh EE et al. N Engl J Med 2023; 388:1465-1477
4 Robert Koch-Institut. Epid Bull 2024;1:311
5 Polkowska-Kramek A et al. Infect Dis Ther 2024;13:845-860
6 Robert Koch-Institut. Epid Bull 2025;41: 3-14
7 Robert Koch-Institut. Epid Bull 2025;31 :14-21
8 Robert Koch-Institut. Epid Bull 2025;15: 3-4
9 Gemeinsamer Bundesausschuss (G-BA): Richtlinie über Schutzimpfungen nach §20i Abs. 1 SGB V (SchutzimpfungsRichtlinie – SI-RL), Fassung vom 05.06.2025, in Kraft getreten am 11.07.2025, veröffentlicht im Bundesanzeiger (BAnz AT 2025)
Wichtig war den Fachautoren, die Perspektive aller einzubeziehen – also auch die der Betroffenen. Daher war eine Patientenvertreterin neben Ärztinnen und Ärzten, Fachpflegenden, Therapeutinnen und Therapeuten wie auch Sozialarbeiterinnen und Sozialarbeitern von Anfang an in die Erstellung eingebunden.
Generell bietet die neue Leitlinie GeriPAIN jetzt sowohl einen interprofessionellen wie auch sektorenübergreifenden Leitfaden für das Schmerzmanagement älterer Menschen – und das für den ambulanten, akutstationären wie auch langzeitstationären Bereich.
Im Dezember 2025 hat die Europäische Kommission Nipocalimab (Imaavy®), einen vollständig humanen monoklonalen Antikörper, der spezifisch auf die IgG-Fc-Bindungsstelle von FcRn abzielt, als Ergänzung der Standardtherapie für die Behandlung der generalisierten Myasthenia gravis (gMG) sowohl für Erwachsene als auch für Jugendliche ab 12 Jahren zugelassen [1].
Programm Referenten
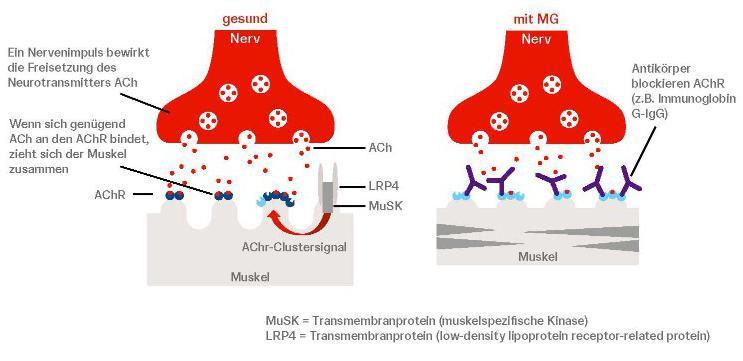
Bei dieser chronischen, unheilbaren neuromuskulären Autoimmunerkrankung bildet das körpereigene Immunsystem Antikörper, die die ACh-Rezeptoren blockieren, verändern oder zerstören. Dadurch wird die Übertragung der Signale von den Nerven an die Muskeln verhindert (Abb. 1). Typische Symptome sind daher eine Schwäche und rasche Ermüdbarkeit der Skelettmuskulatur, die sich zunächst durch Schwierigkeiten beim Kauen, Schlucken und Sprechen
sowie Sehstörungen äußert [2].
Nervenimpuls bewirkt die
Bei bis zu 80 % der Patienten entwickelt sich die Myasthenia gravis meist innerhalb von 2 Jahren nach Ausbruch zu einer generalisierten Form (gMG), bei der weitere Muskelgruppen betroffen sind, z.B. die Gesichts- und pharyngeale Muskulatur, die Atemmuskulatur, die Muskulatur der Extremitäten und rumpfnahe Muskelgruppen. 15 – 20 % der Patienten erleiden mindestens einen schweren, potenziell lebensbedrohlichen Schub
mit MG
Hintergrundinfo Nipocalimab Hintergrundinfo Myasthenia gravis Vorträge Factsheets Key Slides Presseinfo
Bei Myasthenia gravis bildet das körpereigene Immunsystem Antikörper, die die ACh-Rezeptoren blockieren, verändern oder zerstören. Dadurch wird die Übertragung der Signale von den Nerven an die Muskeln verhindert.
– die myasthene Krise. Diese kann eine mechanische Beatmung aufgrund von Atemversagen erforderlich machen [3]. Die meisten Patienten benötigen eine lebenslange Behandlung. Trotz etablierter Standardtherapien besteht noch ein hoher Therapiebedarf. Nipocalimab als neue Option zur Ergänzung der Standardtherapie kann diese Lücke schließen, wie die Ergebnisse der zulassungsrelevanten Studie VIVACITY-MG3 eindrücklich belegen [4, 5]. Nerv
Ein Nervenimpuls bewirkt die Freisetzung des Neurotransmitters ACh gesund mit MG
Wenn sich genügend ACh an den AChR bindet, zieht sich der Muskel zusammen
Antikörper blockieren AChR (z. B. Immunglobulin G-IgG )
AChR - Clustersignal
MuSK = Transmembranprotein (muskelspezifische Kinase)
LRP4 = Transmembranprotein (low-density lipoprotein receptor-related protein)
Abb. 1: Das Krankheitsgeschehen von Myasthenia gravis an der motorischen Endplatte
Abbildung 1: Ursache der Myasthenia gravis ist eine gestörte Impulsübertragung an den Kontaktstellen zwischen Nerven und Muskeln an der motorischen Endplatte. Wenn elektrische Signale einen motorischen Nerv durchlaufen, setzen die Nervenenden den Neurotransmitter Acetylcholin (ACh) frei. Die Transmittermoleküle binden an den Acetylcholinrezeptor (AChR) in der postsynaptischen Membran. Dadurch wird der Muskel aktiviert und zieht sich zusammen (linkes Bild). Bei Myasthenia gravis bildet das körpereigene Immunsystem IgG-Autoantikörper, die die ACh-Rezeptoren blockieren oder zerstören, sodass die Übertragung der Signale von den Nerven an die Muskeln unterbunden wird (rechtes Bild). LRP4 = Low-Density Lipoprotein Receptor-related Protein 4, ein Transmembranprotein, MuSK = muskelspezifische Kinase, ein Transmembranprotein (© Jonson & Johnson).
Anti-AChR- und Anti-MuSK-Antikörper-positive Personen machen 90 % der gesamten Antikörperpositiven gMG-Population aus.8 Bei einer weiteren Form der Myasthenie werden Antikörper gegen das Lipoprotein related protein 4 (LRP4) gebildet.8 Bei sogenannten seronegativen Myasthenien können gar keine Autoantikörper nachgewiesen werden.9 Auch die Thymusdrüse spielt voraussichtlich eine entscheidende Rolle bei der Entstehung der Myasthenia gravis.9
Kontinuierliche Krankheitskontrolle durch FcRn-Blockade bei einer breiten Population von Patienten mit gMG
Nipocalimab ist der erste FcRnBlocker, der sowohl für Erwachsene als auch für Jugendliche ab 12 Jahren mit gMG zugelassen ist, die Antikörper gegen den Acetylcholinrezeptor (AChR) oder die muskelspezifische Tyrosinkinase (MuSK) aufweisen. Anti-AChRund Anti-MuSK-Antikörper-positive Menschen machen ≥90 % der gesamten Antikörper-positiven gMG-Population aus [6].
Programm Referenten
IgG-Autoantikörper, die die neuromuskuläre Übertragung durch Bindung an AChR oder MuSK beeinträchtigen, sind die der Pathogenese der MG zugrundeliegende Ursache. Daher zielt die immunselektive Therapie mit Nipocalimab darauf ab, zirkulierendes Immunglobulin G (IgG) einschließlich pathogener IgG-Autoantikörper zu reduzieren, ohne Auswirkungen auf andere Immunglobuline (IgA, IgE oder IgM) zu generieren.
Nipocalimab bindet spezifisch an den neonatalen Fc-Rezeptor (FcRn), der eine entscheidende Rolle bei der Verlängerung der Halbwertszeit von IgG spielt. Durch die Blockade des FcRn wird der Abbau von IgG gefördert, was zu einer Verringerung von IgG, einschließlich pathogener Autoantikörper, führt (siehe Insert) [7–11]. Der Wirkstoff hat daher das Potenzial, die Krankheitslast bei Autoantikörper- und Alloantikörper-bedingten Krankheiten zu verringern [7].
In einem gesunden Organismus werden IgG-Antikörper in einem IgG-Recycling-Prozess in das Serum zurückgeführt. Dazu bindet der neonatale Fc-Rezeptor (FcRn) an intakte und funktionale Antikörper und transportiert sie an die Zelloberfläche der Zellen, die die Blutgefäße auskleiden. Dieser „Recycling“-Prozess hält den funktionalen Antikörperspiegel im Serum aufrecht. Tritt eine Autoantikörper- oder Alloantikörper-bedingte Krankheit auf, wird auch pathogenes IgG recycelt und seine Konzentration wird im Kreislauf aufrechterhalten, was zur Fortdauer der Krankheit führt [7]. Indem Nipocalimab an FcRn bindet, blockiert es den IgG-Recyclingweg und reduziert die Gesamtmenge zirkulierender IgG-Autoantikörper, die an der Übertragung neuromuskulärer Signale beteiligt sind, sodass sich die krankheitsbedingten neuromuskulären Übertragungsstörungen verringern. Gleichzeitig wird ein Reservoir an Antikörpern für die Funktionsfähigkeit des Immunsystems aufrechterhalten [7].
Hintergrundinfo Nipocalimab Hintergrundinfo Myasthenia gravis Vorträge Factsheets Key Slides Presseinfo
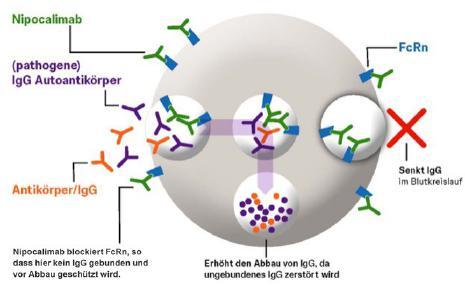
Nipocalimab blockiert FcRn, so dass hier kein IgG gebunden und vor Abbau geschützt wird.
Erhöht den Abbau von IgG, da ungebundenes IgG zerstört wird
Abbildung modifiziert nach Gable K et al. [12].
Senkt IgG im Blutkreislauf
Abb. 1: Wirkmechanismus von Nipocalimab im Rahmen des IgG-Recyclings, modifiziert nach Gable K et al. Antagonism of the Neonatal Fc Receptor as an Emerging Treatment for Myasthenia Gravis.11 FcRn, neonataler Fc Rezeptor; IgG, Immunglobulin G
Forschungsschwerpunkte von Nipocalimab
Autoantikörper-bedingte Krankheiten sind weltweit mit einer erheblichen Morbidität und Mortalität verbunden.12 Sie umfassen eine große und vielfältige Gruppe von bis zu 80 Krankheiten.13 Der FcRnBlocker Nipocalimab wird zurzeit in drei Schlüsselsegmenten im Bereich der Autoimmunerkrankungen untersucht:
• Autoantikörper-bedingte Erkrankungen
(z. B. generalisierte Myasthenia gravis, chronisch entzündliche demyelinisierende Polyneuropathien, autoimmune hämolytische Anämie und idiopathische, entzündliche Myopathien)
◦ In der Phase-III-Zulassungsstudie zur Myasthenia gravis konnte mit Nipocalimab eine IgG-Reduktion von bis zu 75 % erreicht werden.14
Dies belegen die Ergebnisse der zulassungsrelevanten randomisierten, doppelblinden, placebokontrollierten Phase-III-Studie VIVACITY-MG3, die die Wirksamkeit
• Maternal-fetale Erkrankungen, die durch mütterliche Alloantikörper vermittelt werden (z. B. Hämolytische Erkrankung des Fötus und Neugeborenen (HDFN), sogenannte Alloantikörper-bedingte Erkrankungen.)
und Sicherheit von Nipocalimab im Vergleich zu Placebo bei 196 erwachsenen Patienten (davon 153 mit positivem Antiköper-Nachweis gegen den Acetylcholinrezeptor [AChR], die muskelspezifische Tyrosinkinase [MuSK] oder das Low-density Lipoprotein Receptor-related Protein 4 [LRP4]) mit generalisierter Myasthenia gravis untersuchte, die unzureichend auf die Standardtherapie ansprachen* [4]. Die Studienteilnehmer wurden 1:1 auf die Behandlung mit Nipocalimab + Standardtherapie
* Unzureichendes Ansprechen wurde definiert als ein MG-ADL-Gesamtscore ≥6 beim Screening und bei Baseline.
◦ In einer im New England Journal of Medicine veröffentlichten Phase-II-Studie zur HDFN erreichte Nipocalimab eine maximale IgG-Reduktion von 85 %.15
• Prävalente rheumatologische Erkrankungen
(n = 77) oder Placebo + Standardtherapie (n = 76) randomisiert. Nipocalimab (initial 30 mg/kg, danach 15 mg/kg) und Placebo (physiologische Kochsalzlösung) wurden alle 2 Wochen über einen Zeitraum von 24 Wochen als intravenöse Infusion verabreicht. Eine Dosisanpassung von Nipocalimab war nicht zulässig. Während der doppelblinden Phase setzten die Patienten die bestehende Standardtherapie zur Behandlung der Myasthenia gravis unverändert fort (85 % erhielten AChR-Inhibitoren, 66 % Steroide und 54 % nichtsteroidale immunsuppressive Therapien in stabiler Dosierung).
(z. B. rheumatoide Arthritis, Sjögren-Syndrom und systemischer Lupus erythematodes).
◦ In einer auf dem EULAR 2024 und ACR Convergence 2024 vorgestellten Phase-II-Studie zum Sjögren-Syndrom konnten signifikante, dosisabhängige Reduktionen von IgG und Autoantikörpern (AAb) beobachtet werden.16
Teilnehmer, die die doppelblinde Phase abschlossen, konnten im Anschluss im Rahmen einer offenen Verlängerungsphase weiterhin Nipocalimab erhalten [4, 5].
Primärer Wirksamkeitsendpunkt war die Differenz zwischen der Nipocalimab- und Placebo-Gruppe hinsichtlich der mittleren Änderung (Methode der kleinsten Quadrate) des MG-ADL-Gesamtscores** gegenüber dem Ausgangswert, jeweils gemittelt über die Wochen 22,23 und 24 in der Intention-to-treat-Population, bestehend aus Patienten mit positivem Antiköper-Nachweis (AChR+, MuSK+ oder LRP4+).
Als wichtiger sekundärer Endpunkt wurde der Quantitative-Myasthenia-Gravis-(QMG-)Score*** zur Bewertung der Muskelschwäche erhoben [4].
Die Studie zeigte, dass die Patienten, die Nipocalimab plus Standardtherapie erhielten, über einen Zeitraum von 24 Wochen eine bessere Krankheitskontrolle aufwiesen als diejenigen, die Placebo plus Standardtherapie bekamen. Beim primären Endpunkt, der Veränderung des MG-ADL-Gesamtscores gegenüber dem Ausgangswert, ergab sich unter Nipocalimab plus Standardtherapie eine mittlere Verbesserung von –4,70 Punkten versus –3,25 unter Placebo plus Standardtherapie (Differenz = –1,45; 95% KI: –2,38 bis –0,52; p = 0,0024). Auch beim wichtigsten sekundären Endpunkt, dem Quantitative-MyastheniaGravis-(QMG-)Score, war Nipo-
** Die Myasthenia Gravis-specific Activities of Daily Living-Skala (MG-ADL) erfasst die Auswirkungen der Erkrankung auf alltagsrelevante Aktivitäten. Die Skala reicht von 0 bis 24 Punkten, wobei höhere Werte auf schwerere Symptome hinweisen.
*** Ein QMG-Ansprechen war als eine Verringerung des QMG-Gesamtscores um ≥3 Punkte im Vergleich zu Baseline definiert.
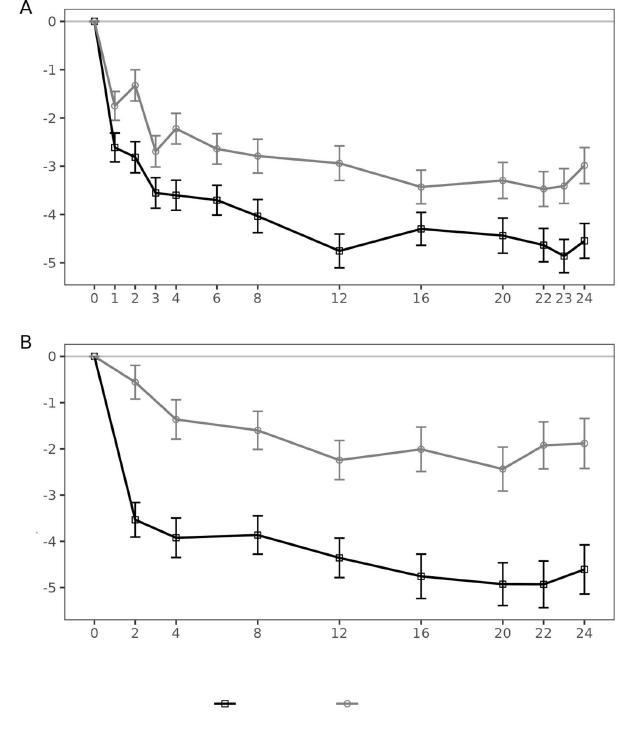
Zeit ab Baseline (Wochen)
Zeit ab Baseline (Wochen) Nipocalimab Placebo
Abbildung 2: Ergebnisse der zulassungsrelevanten Phase-III-Studie VIVACITY-MG3 für den primären Wirksamkeitsendpunkt MG-ADL-Gesamtscore und den sekundären Wirksamkeitsendpunkt QMG-Gesamtscore [1, 4].
calimab überlegen: Unter der Behandlung mit dem FcRn-Blocker sank der QMG-Gesamtscore im Mittel um 4,77 Punkte gegenüber 1,90 Punkten unter Placebo [4] (Abb. 2).
Die Patienten, die in die offene Verlängerungsphase (OLE) eintraten und ihre Behandlung mit Nipocalimab fortsetzten, behielten die Verbesserungen bis zu 20 Monate nach der Nachbeobachtung bei [5]. Damit ist Vivacity-MG3 die längste Zulassungsstudie aller FcRn-Blocker bei Erwachsenen mit gMG, die eine kontinuierliche Krankheitskontrolle über diesen Zeitraum nachweisen konnte.
Die Behandlung mit Nipocalimab führte innerhalb von 2 Wochen nach Behandlungsbeginn zu einer signifikanten raschen Reduktion der Gesamt-IgG-Konzentration im Serum um 75 % gegenüber Baseline, gefolgt von einer anhaltenden Reduktion um 68,98 % gegenüber Baseline von Woche 4 bis Woche 24. Die mittlere prozentuale Veränderung der gesamten Serum-IgG von Baseline bis Woche 24 der aktiven Behandlungsphase war statistisch signifikant (SE: 7,561; 95%-KI: −78,4 bis −59,6) [4].
Ähnliche dosisabhängige Reduktionen wurden in allen IgG-Subklas-
sen (IgG1, IgG2, IgG3 und IgG4) beobachtet [4].
Nipocalimab war während der Studie bei regelmäßiger Gabe gut verträglich und entsprach damit dem Sicherheits- und Verträglichkeitsprofil anderer Studien mit Nipocalimab. Die Gesamtinzidenz unerwünschter Ereignisse war mit 81,6 % im Nipocalimab-Arm und 82,7 % im Placebo-Arm vergleichbar. Schwerwiegende unerwünschte Ereignisse wurden von 9,2 % der Patienten unter Nipocalimab und von 14,3 % der Patienten unter Placebo gemeldet, drei davon mit tödlichem Ausgang (myasthenische Krise unter Nipocalimab, Herzstillstand und Myokardinfarkt unter Placebo [4].
Stuhltest oder Koloskopie? Vor- und Nachteile der Methoden zur Darmkrebsvorsorge
Laut der aktuellen S3-Leitlinie „Kolorektales Karzinom“ der Deutschen Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten (DGVS) senkt die Teilnahme an der gesetzlichen Darmkrebsvorsorge das Erkrankungsrisiko um bis zu 80 %. Die Versicherten haben dabei die Wahl zwischen Koloskopie und Stuhltest:
• Koloskopie: Männer und Frauen haben ab einem Alter von 50 Jahren alle 10 Jahre Anspruch auf eine Darmspiegelung. Bei Vorliegen bestimmter Risikofaktoren ist eine Koloskopie auch schon in jüngerem Alter sinnvoll.
• Immunologischer Stuhltest: Der iFOBT kann alternativ zur Darmspiegelung ab einem Alter von 50 Jahren alle 2 Jahre
Die Myasthenia gravis ist nicht heilbar und die meisten Patienten benötigen aufgrund der sich zunehmend verschlechternden körperlichen Leistungsfähigkeit eine lebenslange Behandlung sowie die Hilfe von Angehörigen und/ oder Pflegekräften. Innovative, zielgerecht in die Pathophysiologie dieser Erkrankung eingreifende Medikamente wie Nipocalimab (Imaavy®) können den Betroffenen neue Hoffnung auf eine verbesserte, kontinuierliche Krankheitskontrolle und eine Steigerung ihrer Lebensqualität geben.
Brigitte Söllner, Erlangen
Literatur
1 Fachinformation Imaavy®; Stand: November 2025
2 Bever CT. Ann Neurol 1983;14:516-519
3 Dresser L et al. J Clin Med 2021;10(11)
4 Antozzi C et al. Lancet Neurol. 2025;24: 105–16 +Supplementaries
5 Antozzi C et al. Long-term safety and efficacy of nipocalimab in generalized myasthenia gravis: Vivacity-MG3 open-label extension phase results. AAN Annual Meeting 2025; Poster #11-022
6 Lazaridis K et al. Front Immunol 2020; 11:212.9
7 Peter HH et al. J Allergy Clin Immunol 2020;146:479-91.e5
8 Ling LE et al. Clin Pharmacol Ther 2019;105:1031-1039
9 Roy S et al. Am J Obstet Gynecol 2019;220:498.e1-e9
10 Patel DD et al. J Allergy Clin Immunol 2020;146:467-478
11 Roopenian DC et al. Nat Rev Immunol 2007;7:715-725
12 Gable KL et al. Front Immunol 2019; 10:3052
durchgeführt werden, kommt davor aber auch schon im Rahmen des allgemeinen Gesundheits-Check-ups zum Einsatz.
• Beide Verfahren verfolgen ein gemeinsames Ziel: Adenome – also Krebsvorstufen – aufzuspüren und rechtzeitig zu entfernen, bevor sie entarten.
Stuhltest: niedrigschwellig, aber nur bei Regelmäßigkeit effektiv
Darmspiegelung: der Goldstandard mit therapeutischem Potenzial
Bei der Koloskopie wird der gesamte Dickdarm mithilfe eines flexiblen Endoskops untersucht. Sie erfolgt auf Wunsch in Kurznarkose und dauert etwa 20 – 30 Minuten. Die Koloskopie erkennt nicht nur frühe Tumoren, sie verhindert auch aktiv Krebs, weil Polypen während der Untersuchung schmerzfrei und risikoarm entfernen werden können. Laut aktueller S3-Leitlinie lässt sich durch die Koloskopie das Risiko, an Darmkrebs zu erkranken, um bis zu 80 % senken. Ihre Einmaligkeit bei hoher Wirksamkeit macht sie zur effektivsten Vorsorgemaßnahme.
F. S.
Der immunologische Stuhltest ist auch zu Hause durchführbar. Er ist einfach, aber sensitiv genug, um Blut im Stuhl zu erkennen, das von kleinen Tumoren oder Adenomen stammen kann. Der Test ist damit auch für Menschen geeignet, die Hemmungen vor einer Darmspiegelung haben. Der iFOBT erreicht eine Sensitivität von rund 30 –40 % für fortgeschrittene Adenome und über 70 % für invasive Karzinome, sollte dafür aber regelmäßig wiederholt werden. Ein positives Testergebnis erfordert zwingend eine nachfolgende Darmspiegelung zur Abklärung.
Wie aktuelle, auf einem Fachpressegespräch von Novartis vorgestellte Daten der Phase-III-Studien REMIX-1 und REMIX-2 zeigen, kann der hochselektive, oral zu verabreichende BTK-Inhibitor Remibrutinib bereits ab Woche 1 zu signifikanten Verbesserungen der Lebensqualität, des Schlafs und der Alltagsaktivität von Patienten mit chronischer spontaner Urtikaria (csU) führen [1]. Diese systemische Autoimmunerkrankung äußert sich durch plötzlich auftretende Angioödeme und/oder Quaddeln [2], die die Betroffenen stark belasten und in ihrer Lebensqualität stark einschränken können [3], weshalb die Patienten häufig auch psychische Begleiterkrankungen wie Depressionen oder Angstzustände entwickeln [4].
Hoher Leidensdruck aufgrund verzögerter Diagnose und unzureichender Symptomkontrolle
Der ausgeprägte Leidensdruck erfordert eine frühzeitige Diagnosestellung und wirksame Therapieansätze. Laut Urticaria-VoicesStudie verzögert sich die Diagnose aber im Durchschnitt um rund 2 Jahre und rund 80 % der Patienten berichten trotz Behandlung von einer unzureichenden Symptomkon-
trolle [5]. Hinzu kommen deutliche Einschränkungen des psychischen Wohlbefindens, des sozialen Lebens sowie der alltäglichen Aktivitäten, wobei auch Schlafstörungen zu den am stärksten belastenden Auswirkungen der csU gehören [5].
Deutliche Reduktion des Urtikaria-Aktivitäts-Scores
In der Analyse von insgesamt 912 Patienten, die an den beiden identisch konzipierten, randomisierten, doppelblinden und placebokontrollierten Parallelgruppenstudien REMIX-1 (n = 740) und REMIX-2 (n = 455) teilnahmen, wobei 606 mit Remibrutinib behandelt wurden und 306 Placebo erhielten [6, 7], zeigte sich bereits ab Woche 1 eine Reduktion des wöchentlichen Urtikaria-Aktivitäts-Scores (UAS7) unter Remibrutinib 25 mg zweimal täglich gegenüber Placebo (18,8 vs. 26,1). Bis Woche 24 verbesserte sich der Score weiter auf 8,3 versus 14,2 unter Placebo [1]. Mehr als ein Drittel der Patienten (35,6 %) erreichte eine vollständige Symptomfreiheit (UAS7 = 0), verglichen mit 18 % unter Placebo [1].
Zu Studienbeginn hatten beide Gruppen einen stark beeinträchtigten DLQI-Score von etwa 14 Punkten. Unter Remibrutinib ver-
besserte sich die dermatologische Lebensqualität schneller als unter Placebo bereits in Woche 4 (5,3 vs. 8,9 Punkte) und bis Woche 24 von „einer sehr großen“ auf „eine geringe Auswirkung“ (4,4 vs. 6,7 Punkte). Die starke Korrelation zwischen Urtikaria-Aktivität und Lebensqualität (0,72) belegt den direkten Zusammenhang zwischen Symptomkontrolle und Patientenwohlbefinden [1].
Bessere Symptomkontrolle wirkt sich positiv auf die Schlafqualität und Alltagsfunktion aus
Die anfangs stark beeinträchtigte Schlafqualität in beiden Gruppen (SIS7: 12,2 Punkte) verbesserte sich unter Remibrutinib bereits ab Woche 1 (7,4 vs. 10,3 unter Placebo) mit anhaltender Reduktion bis Woche 24 (2,9 vs. 4,8 Punkte) [1]. Die starke Korrelation zwischen Urtikaria-Aktivität und Schlafbeeinträchtigung (0,80) macht deutlich, dass eine bessere Symptomkontrolle direkt zu erholsamerem Schlaf führen kann [1].
Unter der Therapie mit Remibrutinib verbesserte sich auch die eingeschränkt Alltagsfunktion (AIS7; Ausgangswert ~12,7 Punkte) bereits ab Woche 1 auf 7,3 vs. 10,4 Punkte unter Placebo mit weiterer Reduktion bis Woche 24 (3,3 vs. 5,1 Punkte). Der ausgeprägte
Remibrutinib ist ein hochselektiver, kovalenter, oral zu verabreichender Bruton-Tyrosinkinase (BTK)-Inhibitor. Die Aktivierung der BTK-Signalkaskade führt bei chronischer spontaner Urtikaria (cSU) zur Ausschüttung von Histamin, das Quaddeln und Schwellungen verursacht. Durch die gezielte Blockade von BTK wird dieser Vorgang verhindert [9, 10, 11]. In Phase-III-Studien zeigte Remibrutinib bei Patienten mit mittelschwerer bis schwerer csU eine signifikante Verbesserung der Symptomkontrolle (p < 0,001) [8].
Remibrutinib (Handelsname Rhapsido) hat im September 2025 die Zulassung der US-Gesundheitsbehörde FDA für Erwachsene mit chronischer spontaner Urtikaria (CSU) erhalten, die auf Antihistaminika nicht ansprechen. Die EU-Zulassung steht noch aus.
Zusammenhang zwischen Urtikaria-Aktivität und Aktivitätsbeeinträchtigung (0,84) zeigt, dass eine Symptomkontrolle die Bewältigung des Alltags unmittelbar verbessern kann [1].
Günstiges Sicherheitsprofil
Remibrutinib zeigte in beiden Studien eine gute Verträglichkeit und ein günstiges Sicherheitsprofil, wobei die Ergebnisse der Leberfunktionstests bis in Woche 52 unauffällig waren [8]. Die am häufigsten beobachteten unerwünschten Ereignisse in den REMIX-Studien waren Infektionen der Atemwege (COVID-19 und Nasopharyngitis, beide vergleichbar mit Placebo).
Der hochselektive Bruton-Tyrosinkinase (BTK)-Inhibitor Remibrutinib erwies sich in den Phase-IIIStudien REMIX-1 und REMIX-2 als eine wirksame Behandlungsoption für csU-Patienten mit unzureichender Symptomkontrolle unter H1-Antihistaminika. Die frühe Wirksamkeit ab Woche 1 und die starken Korrelationen mit klinisch
bedeutsamen Parametern unterstreichen das Potenzial für eine rasche und umfassende Verbesserung der Krankheitslast [8].
Brigitte Söllner, Erlangen
Die Europäische Kommission hat Pirtobrutinib (Jaypirca®) als ersten reversiblen Bruton-Tyrosinkinase-Inhibitor (BTKi) jetzt auch zur Monotherapie von Erwachsenen mit rezidivierter oder refraktärer chronisch lymphatischer Leukämie (r/r CLL) nach BTKi-Vorbehandlung zugelassen [1]. Voraussetzung waren die Ergebnisse der Phase-III-Studie BRUIN CLL-321, in der mit Pirtobrutinib das progressionsfreie Überleben der Patienten bei guter Verträglichkeit signifikant verlängert werden konnte [2].
Die CLL wird der Gruppe indolenter B-Zell-Lymphome zugeordnet [3]. In Deutschland ist sie die häufigste Form von Blutkrebs bei Erwachsenen. Sowohl in der Erstals auch in der Zweitlinientherapie werden heute meist Chemotherapie-freie, zielgerichtete Behandlungsoptionen eingesetzt [3]. Einer dieser Ansätze beruht auf der Hemmung der für die Regulation von Proliferation und Überleben gesunder und maligner B-Zellen wichtigen Bruton-Tyrosinkinase (BTK) [3, 4].
Literatur
1 Lebwohl M et al. ACAAI 2025, Poster R093
2 AWMF. https://register.awmf.org/assets/ guidelines/013-028l_S3_KlassifikationDiagnostik-Therapie-Urtikaria_2022-04. pdf
3 Fricke J et al. Allergy 2020;75:423-432
4 Peters EMJ. J Dtsch Dermatol Ges 2016; 14:233-252
5 Weller K et al. Dermatol Ther (Heidelb) 2025;15:747-761
6 Novartis Pharmaceuticals. https://clinicaltrials.gov/study/NCT05030311
7 Novartis Pharmaceuticals. https://clinicaltrials.gov/study/NCT05032157
8 Metz M et al. EAACI 2024. Abstract
9Angst D et al. J Med Chem 2020;63:51025118
10 Powell RJ et al. Clin Exp Allergy 2015;45:547-565
11 Maurer M et al. J Allergy Clin Immunol 2022;150:1498-1506.e2
BRUIN CLL-321 – die einzige Phase-III-Studie mit 100 % BTKivorbehandelten Patienten
Grundlage der Zulassungserweiterung sind die Ergebnisse der randomisierten Phase-III-Studie BRUIN CLL-321 [2]. Sie ist die einzige r/r CLL-Studie, in der ausschließlich mit kovalenten BTKi-vorbehandelte Patienten nach rezidivierendem oder refraktärem Verlauf untersucht wurden [1, 2]. Stratifiziert nach den Parametern einer vorliegenden 17p-Deletion und einer BCL-2-Inhibitor-Vorbehandlung erhielten die 238 Studienteilnehmer 1:1 randomisiert entweder die Pirtobrutinib-
Monotherapie (200 mg einmal täglich, n = 119) oder eine kombinierte Chemo-Immuntherapie aus Idelalisib plus Rituximab (Idela®; n = 82) oder Bendamustin plus Rituximab (BR; n = 37) nach Wahl des Prüfarztes. Bei einem bestätigten Progress unter den Vergleichstherapien war ein Crossover aus dem Kontroll- in den Pirtobrutinib-Verumarm möglich [2].
Signifikante Verlängerung des PFS und der Zeit bis zur nächsten Therapie
Primärer Studienendpunkt war das progressionsfreie Überleben (PFS). Nach einer medianen Nachbeobachtungszeit von ca. 19 Monaten betrug das es unter Pirtobrutinib 14,0 Monate gegenüber 8,7 Monaten im Kontrollarm (HR: 0,54; 95%-KI: 0,39 – 0,75; p = 0,0002). Die Therapie mit Pirtobrutinib reduzierte das Risiko für Progress oder Tod um 46 % [2]. Ein sekundärer Endpunkt war die Zeit bis zur nächsten Behandlung oder bis zum Tod (TTNT). Die TTNT betrug unter Pirtobrutinib im Median 2 Jahre und lag hiermit etwa 13 Monate über der TTNT im Kontrollarm (TTNT: 24,0 vs. 10,9 Monate; HR: 0,37; 95%-KI
0,25 – 0,52; p < 0,0001). Damit konnte das Risiko, eine neue Therapie beginnen zu müssen oder zu versterben, mit Pirtobrutinib um 63 % verringert werden [2]. Wichtig zu erwähnen ist, dass ein hoher Anteil der Patienten charakteristische Merkmale eines aggressiven Krankheitsverlaufs aufwies, einschließlich der Hochrisikofaktoren TP53-Mutation und/oder 17p-Deletion, sowie einen unmutierten IGHV-Status und komplexen Karyotyp. Die Patienten waren stark vorbehandelt und hatten im Median 3 Vortherapien [2]. Dies unterstreicht die Wirksamkeit von Pirtobrutinib auch bei ungünstiger Prognose.
Gute Verträglichkeit der Pirtobrutinib-Therapie
In der BRUIN CLL-321-Studie waren die meisten der beobachteten unerwünschten Ereignisse (UE) vom Grad 1 oder Grad 2. Nur 5 % der Patienten brachen die Therapie aufgrund von behandlungsbedingten Nebenwirkungen ab. Blutungen und kardiovaskuläre UEs traten nur vereinzelt auf [2]. Das Nebenwirkungsprofil war konsistent mit den Daten der Phase-I/II-BRUIN-Studie [5, 6].
Innovativer Wirkmechanismus ermöglicht Fortsetzung der BTKHemmung bei rezidivierendem und refraktärem Verlauf
Unter der initialen CLL-Therapie mit kovalenten BTKi können Resistenzmutationen auftreten [7]. Als erster Vertreter der Substanzklasse nicht-kovalenter BTKi bindet Pirtobrutinib reversibel und unabhängig von bekannten Mutationen – wie den C481-Mutationen – in der ATP-Bindungstasche des BTKProteins [1, 5, 8]. Hierdurch können Resistenzen überwunden werden [5, 9]. Bei der r/r CLL kann mit Pirtobrutinib im post-BTKi-Setting so der BTK-Signalweg weiterhin blockiert werden [5, 8, 9]. Die Patienten können daher die BTKi-Therapie mit gewohntem und einfachem Dosierschema (200 mg täglich oral) fortführen [1].
Fabian Sandner, Nürnberg
Literatur
1 Fachinformation Jaypirca®, aktueller Stand
2 Sharman JP et al. 886 BRUIN CLL-321: Randomized phase III trial of pirtobrutinib versus idelalisib plus rituximab (Idela®) or bendamustine plus rituximab (BR) in BTK inhibitor pretreated chronic lymphocytic leukemia/small lymphocytic lymphoma. Abstract presented at: American Society of Hematology Annual Meeting and Exposition; December 9, 2024; San Diego, CA. Session 642
3 Wendtner CM et al. Chronische Lymphatische Leukämie (CLL). Onkopedia Leitlinie, Stand: September 2024
4 Tasso B et al. Molecules 2021;26:7411
5 Mato AR et al. Lancet 2021;397:892-901
6 Mato AR et al. N Engl J Med 2023;389: 33-44
7 Cheah CY et al. Ann Oncol 2015;26: 1175-1179
8 Aslan B et al. Blood Cancer J 2022;12:80
9 Gu D et al. J Hematol Oncol 2021;14:40
IQWiG sieht bei Lecanemab keinen Zusatznutzen – eine Stellungnahme von Eisai
Das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) hat am 01.12.2025 seinen Bericht zur Bewertung des Zusatznutzens von Leqembi® veröffentlicht. Eisai ist zutiefst enttäuscht über die Bewertung des IQWiG, laut der für Leqembi®, der ersten in Deutschland erhältlichen Therapie der frühen AlzheimerKrankheit, die auf zugrundeliegende Krankheitsprozesse abzielt [1], im Vergleich zu der vom Gemeinsamen Bundesausschuss (GBA) festgelegten zweckmäßigen Vergleichstherapie (ZVT) kein Zusatznutzen belegt sei [2]. Eisai widerspricht dieser Sichtweise entschieden und betont die für Patienten direkt relevanten und bedeutsamen Ergebnisse zur Wirksamkeit. Die Bewertung des IQWiG ist ein Vorschlag an den G-BA, dessen Beschlussfassung im Februar 2026 erwartet wird.
Leqembi® wird angewendet zur Behandlung erwachsener Patienten mit klinisch diagnostizierter leichter kognitiver Störung (mild cognitive impairment, MCI) und leichter Demenz aufgrund der AlzheimerKrankheit (zusammengenommen frühe Alzheimer-Krankheit) mit bestätigter Amyloid-Pathologie, die Apolipoprotein E ε4 (ApoE ε4)-Nichtträger oder heterozygote ApoE ε4-Träger sind [3].
Effektiv in allen Wirksamkeitsendpunkten
Grundlage der Zulassung von Lecanemab war die internationale
randomisierte, kontrollierte Studie Clarity AD, an der 1.795 Erwachsene (Einschlusskriterium Alter: 50–90 Jahre) mit früher symptomatischer Alzheimer-Krankheit teilnahmen. Für den primären Endpunkt, den CDR-SB (Clinical Dementia Rating – Sum of Boxes), der Kognition und Alltagsfunktion bei Patienten anhand verschiedener Domänen abbildet, zeigte sich eine Verlangsamung der Krankheitsprogression um 27 % im Vergleich zur Placebo-Gruppe nach 18 Monaten (Differenz: –0,45 Punkte; p < 0.001). Alle sekundären Endpunkte zeigten ebenfalls statistisch signifikante Unterschiede zu Gunsten von Lecanemab gegenüber Placebo [4].
Für die EU-Zulassung erfolgte eine Post-hoc-Analyse, bei der nur die Gruppe der ApoE ε4-Nichtträger und heterozygoten ApoE ε4Träger (n = 1.521) berücksichtigt wurden [5]. Die Ergebnisse der Wirksamkeitsendpunkte in dieser Subpopulation waren konsistent mit denen der Gesamtkohorte der Clarity AD-Studie. Für den primären Endpunkt CDR-SB zeigte sich in dieser Subpopulation eine Verlangsamung der Krankheitsprogression um 31 % im Vergleich zur Placebo-Gruppe nach 18 Monaten (Differenz: –0,54 Punkte; p = 0,00001) [5].
IQWiG-Bewertung ignoriert nachgewiesene Wirksamkeit
Das IQWiG hat die Studienpopulation der Clarity AD nach Krankheitsstadien aufgetrennt und jeweils gegen eine unterschiedliche ZVT bewertet (MCI aufgrund der Alzheimer-Krankheit: beobachtendes Abwarten; leichte AlzheimerDemenz: symptomatische Therapie mit Acetylcholinesterase-
Inhibitor [AChEI]). Die Auftrennung wird dem als Kontinuum verstandenen Anwendungsgebiet der frühen Alzheimer-Krankheit nicht gerecht. Zudem berücksichtigt das IQWiG in seinen Analysen lediglich eine Minderheit der in Clarity AD untersuchten Patienten. Das IQWiG betrachtet ausschließlich MCI-Patienten, die keine Antidementiva erhalten haben und Patienten mit leichter AlzheimerDemenz, die mit AChEI behandelt wurden [2]. Dies ignoriert die Versorgungswirklichkeit in Deutschland und lässt die bestverfügbare und umfangreiche Evidenz in dieser Indikation außen vor.
In dem von Eisai vorgelegten Nutzendossier [6] wurde die Ableitung des Zusatznutzens für Leqembi® basierend auf der Gesamtpopulation der Studie Clarity AD vorgenommen. Das IQWiG kritisiert, dass dieses Vorgehen nicht die Einschränkungen der Zulassung sowie die vom G-BA festgelegte Auftrennung der Studienpopulation und der jeweils dafür bestimmten ZVT berücksichtigt [2]. Eisai hat hingegen im Nutzendossier gezeigt, dass die Auftrennung der Studienpopulation sowie der Ausschluss von Patienten mit homozygotem ApoE ε4-Trägerstatus oder der Verwendung von Antikoagulanzien auch aus statistischer Sicht nicht notwendig sind. Die Betrachtung der Gesamtpopulation erlaubt somit die verlässlichste Interpretation der Studiendaten. Die Vorgehensweise im Dossier wird aus Sicht von Eisai weiterhin als sachgerechte Umsetzung der Vorgaben zur ZVT durch den G-BA betrachtet. Zudem wird die deutsche Versorgungswirklichkeit sowohl in Clarity AD als auch im Nutzendossier gut abgebildet [4, 6].
Eisai hat in seinem Dossier Informationen zu Wirksamkeits- und
insbesondere Verträglichkeitsendpunkten in großer Detailtiefe und den Vorgaben des G-BA entsprechend vorgelegt [6]. Die Interpretation dieser Daten durch das IQWiG hält Eisai für nicht angemessen.
Zusammenfassend sieht Eisai die wesentlichen Voraussetzungen an die Ableitung eines Zusatznutzens erfüllt. Eisai ist der Auffassung, dass das IQWiG es versäumt hat, die patientenrelevanten Vorteile von Leqembi® angemessen zu berücksichtigen. Der innovative Charakter von Leqembi® in einem klinischen Umfeld mit einem hohen ungedeckten therapeutischen Bedarf wird nicht ausreichend gewürdigt.
Die endgültige Entscheidung über den Zusatznutzen von Leqembi® liegt beim G-BA. Neben dem Vorschlag im Bericht des IQWiG wird der G-BA das Nutzendossier von Eisai, die schriftlichen Stellungnahmen und die mündliche Anhörung, in die auch klinische
Experten und Patientenvertreter eingebunden sind, in seine Entscheidung über den Zusatznutzen einbeziehen. Eisai ist zuversichtlich, dass der G-BA hierbei einen flexibleren und patientenorientierten Ansatz zugrundelegen wird, welcher sowohl die Evidenz des klinischen Nutzens von Leqembi® als auch die Bedürfnisse von Patienten mit früher AlzheimerKrankheit umsichtig einbeziehen wird.
gemeinsame Zeit mit dem geliebten Menschen“, kommentiert Prof. Dr. Jörg Schulz, Direktor der Klinik für Neurologie am Universitätsklinikum Aachen.
Quellen
1 1 G-BA. Tragende Gründe zum Beschluss des Gemeinsamen Bundesausschusses über eine Änderung der Arzneimittel-Richtlinie: Anlage III (Verordnungseinschränkungen und -ausschlüsse – Nummer 10a (Lecanemab) Stand: 20. November 2025. https://www.g-ba.de/ downloads/40-268-12081/2025-11-20_ AM-RL-III-Nr10a-Antidementiva-Lecanemab_TrG.pdf
2 IQWiG. Lecanemab (frühe AlzheimerKrankheit) Nutzenbewertung gemäß § 35a SGB V. Projekt: A25-111. Stand: 27.11.2025. www.g-ba.de/downloads/92975-9111/2025-12_01_Nutzenbewertung-IQWiG_Lecanemab_D-1054.pdf
3 Fachinformation Leqembi®; Stand: September 2025
4 van Dyck CH et al. New Engl J Med 2023;388:9-21
5 Froelich L et al. DGPPN-Kongress 2024; Poster 17

„Mit Leqembi® als krankheitsmodifizierende Therapie haben wir zum ersten Mal die Möglichkeit, das Fortschreiten der Erkrankung tatsächlich zu verlangsamen. Nach mehr als 20 Jahren intensiver Forschung mit vielen Rückschlägen ist dies ein großer medizinischer Meilenstein und gibt den Betroffenen neue Hoffnung im Umgang mit der Erkrankung. Für die Patienten bedeutet dies länger aktiv am Leben teilhaben zu können und für Angehörige kostbare
6 Eisai GmbH. Dossier zur Nutzenbewertung gemäß § 35a SGB V. Lecanemab (LEQEMBI®). www.g-ba.de/bewertungs verfahren/nutzenbewertung/1257/


CME-zertifizierte Online-Fortbildungen zum Thema Alzheimer-Krankheit

Machen Sie mit und melden Sie sich an!
Machen Sie mit und melden Sie sich an!
CME-zertifizierte Online-Fortbildungen zum Thema Alzheimer-Krankheit
Alzheimer-Krankheit
Scannen Sie zur Registrierung den QR-Code oder geben Sie folgenden Link in Ihrem Webbrowser ein: https://www.eisaipro.eu/de-de/neurology/fortbildung/veranstaltungen/neuronar/anmeldung
Machen Sie mit und melden Sie sich an!
Scannen Sie zur Registrierung den QR-Code oder geben Sie folgenden Link in Ihrem Webbrowser ein: https://www.eisaipro.eu/de-de/neurology/fortbildung/veranstaltungen/neuronar/anmeldung
Termine und Themen#:
Termine und Themen#:
Termine und Themen#:
25.02.26 Alzheimer-Krankheit: Vom Molekül zu Netzwerken zur Therapie
Prof. Marc Aurel Busche, Basel
25.02.26 Alzheimer-Krankheit: Vom Molekül zu Netzwerken zur Therapie
25.02.26 Alzheimer-Krankheit: Vom Molekül zu Netzwerken zur Therapie
Prof. Marc Aurel Busche, Basel
Prof. Marc Aurel Busche, Basel
Scannen Sie zur Registrierung den QR-Code oder geben Sie folgenden Link in Ihrem Webbrowser ein: https://www.eisaipro.eu/de-de/neurology/fortbildung/veranstaltungen/neuronar/anmeldung # Änderungen vorbehalten
25.03.26 Tempus fugit – Erkenntnisse aus den ersten 6 Monaten mit Lecanemab
Prof. Frank Jessen, Köln
25.03.26 Tempus fugit – Erkenntnisse aus den ersten 6 Monaten mit Lecanemab
25.03.26 Tempus fugit – Erkenntnisse aus den ersten 6 Monaten mit Lecanemab
Prof. Frank Jessen, Köln
Prof. Frank Jessen, Köln
# Änderungen vorbehalten
Informationen zum Datenschutz bei Eisai erhalten Sie in unserer Datenschutzerklärung unter https://www.eisai.de/datenschutz.
Änderungen vorbehalten
Die Einladung für diese von der Eisai GmbH organisierte Online-Fortbildungsveranstaltung kann nur für fachliche Teilnehmer:innen ausgesprochen werden.
kann nur für fachliche Teilnehmer:innen ausgesprochen werden.
DE-NEUR-26-00017 Biogen-280961
Primär biliäre Cholangitis: Lyvdelzi® löst Seladelpar Gilead ab
Im Februar 2025 wurde Seladelpar zunächst unter der Bezeichnung „Seladelpar Gilead“ zur Behandlung der primär biliären Cholangitis (PBC) zugelassen. Es ist in Kombination mit Ursodeoxycholsäure (UDCA) bei Erwachsenen indiziert, die nicht ausreichend auf UDCA alleine ansprechen, oder als Monotherapie bei Patienten, die UDCA nicht vertragen. Seit Mai 2025 ist das Medikament unter dem neuen Handelsnamen Lyvdelzi® verfügbar. Seladelpar Gilead wurde „außer Vertrieb“ (AV) gemeldet, ist aber weiterhin abgabefähig. Da Vorgänger- und Nachfolgeartikel der Aut-idemRegelung entsprechen, sind diese uneingeschränkt austauschbar.
Signifikante Verringerung des Pruritus
Die Wirksamkeit und Sicherheit von Seladelpar wurden in der randomisierten, doppelblinden, placebokontrollierten, 12-monatigen Phase-III-Studie RESPONSE bei PBC-Patienten untersucht. In der Zulassungsstudie wurde der primäre Endpunkt, das kombinierte biochemische Ansprechen (Alkalische-Phosphatase [ALP] <1,67 × ULN, Gesamtbilirubin
Seladelpar
≤ULN und ALP-Abnahme ≥15 %) in Monat 12, bei 61,7 % der mit Seladelpar behandelten Patienten erreicht. In der Placebo-Gruppe war das bei 20,0 % der Fall (95%-KI: 27,7 – 53,4; p < 0,001). Zu einer Normalisierung des ALP-Spiegels in Monat 12 kam es bei 25,0 % der Patienten in der Verum-Gruppe, in der Placebogruppe bei 0,0 % (95%-KI: 18,3 – 33,2; p < 0,001).
Seladelpar führte nach 6 Monaten zu einer signifikanten Reduktion des Juckreizes bei Patienten mit moderatem bis schwerem Pruritus (NRS ≥4) gegenüber Placebo (–3,2 versus –1,7; p = 0,005; 95%-KI: –2,5 bis –0,5).
Unerwünschte Ereignisse traten bei 86,7 % der Patienten unter Seladelpar und bei 84,6 % der Patienten unter Placebo auf. Unerwünschte Ereignisse, die zu einem Behandlungsabbruch führten, waren in beiden Studienarmen selten (bei 3,1 % unter Seladelpar versus 4,6 % unter Placebo). Am häufigsten (bei ≥10 % in beiden Gruppen) waren eine COVID19-Erkrankung und Pruritus. In weiteren klinischen Studien waren die am häufigsten berichteten Nebenwirkungen Abdominalschmerz (11,1 %), Kopfschmerzen (7,2 %), Übelkeit (6,5 %) und aufgeblähter Bauch (3,9 %). Diese Nebenwirkungen waren nicht schwerwiegend und führten nicht zu einem Abbruch der Behandlung mit Seladelpar.
F. S.
Seladelpar (Lyvdelzi®) ist ein selektiver Agonist des Peroxisom-Proliferator-aktivierten Rezeptors delta (PPARδ). PPARδ ist ein Kernrezeptor, der in der Leber und in anderen Geweben exprimiert wird. Die Aktivierung von PPARδ verringert die Gallensäuresynthese in der Leber, was zu einer niedrigeren Exposition gegenüber Gallensäure in der Leber und zu verminderten Konzentrationen der zirkulierenden Gallensäure führt. Die empfohlene Dosis Seladelpar beträgt 10 mg einmal täglich [2].
Bei CED immer die Leber mitdenken
Die Leber rückt immer weiter in das Blickfeld von Gastroenterologen, die sich mit chronisch entzündlichen Darmerkrankungen (CED) wie Colitis ulcerosa (CU) und Morbus Crohn (MC) beschäftigen. Dies wurde auch auf der Tagung der Deutschen Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten deutlich: Jeder Zweite mit einer CED leidet an extraintestinalen Manifestationen (EIM). Häufig sind die Leber und die Gallenblase betroffen. Bis zu 8 % der Patienten mit CU leiden gleichzeitig an einer primär sklerosierenden Cholangitis (PSC). Auch medikamentöse CED-Therapien können sich negativ auf die Lebergesundheit auswirken. Die klinische Relevanz der DarmLeber-Achse, das diagnostische Handwerkszeug, Routineuntersuchungen sowie therapeutische Ansätze bei CED und Lebererkrankungen waren daher Themen auf einem von der Dr. Falk Pharma GmbH organisierten Satellitensymposium.
Hepatobiliäre Komorbiditäten sind häufige extraintestinale Manifestationen
Bei Morbus Crohn und Colitis ulcerosa ist häufig nicht nur der Verdauungstrakt von entzündlichen Veränderungen betroffen. Bei CED können beispielsweise auch Organe außerhalb des Gastrointestinaltrakts, wie z.B. die Augen, die Haut oder die Gelenke betroffen sein. Häufig werden die
Leber und die Gallenwege in Mitleidenschaft gezogen. Diese hepatobiliären Komorbiditäten zählen sogar zu den häufigsten extraintestinalen Manifestationen. Bei etwa einem Drittel der Patienten mit CED sind die Leberwerte erhöht, 5 % der Betroffenen entwickeln eine chronische Lebererkrankung. Bleiben Lebererkrankungen bei CED-Patienten unentdeckt, kann dies zu Komplikationen führen. Eine regelmäßige Überwachung der Leber ist deshalb bei chronisch entzündlichen Darmerkrankungen besonders wichtig. Ein Zusammenhang zwischen Lebererkrankungen und CED kann in einer eng verbundenen Pathogenese begründet sein. So kann es bei primär sklerosierender Cholangitis (PSC), IgG4-bedingter Cholangitis und Autoimmunhepatitis (AIH) einen gemeinsamen Autoimmunhintergrund mit CED geben. Aber auch eine gemeinsame Stoffwechselstörung (nicht-alkoholische Fettlebererkrankung bzw. Cholelithiasis) oder eine Medikamenteninduzierte Toxizität kann Basis für die Erkrankung beider Organsysteme sein.
PSC als Muster-Ko-Morbidität bei CED
Der Entstehung einer primär sklerosierende Cholangitis liegt offenbar eine komplexe Interaktion aus genetischen und Umweltfaktoren zugrunde. Die genaue Ursache der Erkrankung ist aber nicht hinreichend bekannt. Von der chronisch cholestatischen Lebererkrankung sind sowohl die intra- als auch die extrahepatischen Gallengänge betroffen. Ein Großteil (ca. 70 %) der Menschen mit einer PSC leidet gleichzeitig an einer CED, meist an einer Colitis ulcerosa. Umge-
kehrt wird bei schätzungsweise 3 % der Patienten mit MC und bei bis zu 8 % derer mit CU auch eine PSC festgestellt. Die überwiegende Mehrzahl der Patienten mit einer PSC zeigt laborchemisch eine erhöhte alkalische Phosphatase (AP). Eine erhöhte γ-GlutamylTransferase (γ-GT) ergänzt oft das typische cholestatische Laborprofil, die Serumtransaminasen sind meist nur diskret erhöht. Das Serum-Bilirubin ist bei der Erstdiagnose der PSC überwiegend normal und erhöht sich erst bei zunehmendem Schweregrad der Erkrankung. In der klinischen Untersuchung zeigt sich vor allem eine Hepatomegalie bzw. Splenomegalie. Da die laborchemischen Parameter keine eindeutigen Hinweise auf die PSC liefern, sollte die Diagnose durch eine Magnetresonanz-Cholangiopankreatikographie (MRCP) erfolgen.
Erhöhtes Risiko für ein kolorektales Karzinom
Im Vergleich zur isolierten CED ist beim Vorliegen einer PSC-CED das Risiko für ein kolorektales Karzinom erhöht. Aus diesem Grund hat die PSC-Diagnose einen wichtigen diagnostischen Wert. Liegen sowohl hepatobiliäre Erkrankungen als auch chronisch entzündliche Darmerkrankungen vor, sollte das Kolonkarzinom-Screening früher begonnen und jährlich durchgeführt werden. Das Vorliegen einer PSC bei CED beeinflusst also die Prognose erheblich. Weitere Komplikationen können eine Cholangitis und Cholezystolithiasis, Cholangiokarzinome, Osteoporose, Vitaminmangel und Steatorrhö sein.
CED-Therapien und deren Einfluss auf die Leber
Neben Ko-Morbiditäten wie der PSC können auch die medikamentösen Behandlungsoptionen der CED ein erhöhtes Risiko für eine Hepatotoxizität tragen. Deshalb empfiehlt die European Crohn’s and Colitis Organization (ECCO) alle 1 – 3 Monate eine Leberwertbestimmung.
5-ASA (5-Aminosalicylsäure)Präparate gehen selten mit einer Drug-Induced Liver Injury (DILI) einher. Allerdings werden unter der Therapie mit Methotrexat bei rund 10 % der Patienten mit CED erhöhte Transaminasen-Werte beobachtet. Auch bei manchen Biologika wurden Fälle von Hepatotoxizität beschrieben. In der klinischen Praxis sollte daher ein gezieltes Monitoring angestrebt werden, um DILI frühzeitig zu erkennen.
Derzeit existiert keine Standardtherapie der PSC. Die Off-label-Behandlung mit Ursodesoxycholsäure (UDCA) in mittlerer Dosierung kann zu einer Verbesserung der Laborparameter führen. Abhängig von Symptomen und Komplikationen können zusätzliche medikamentöse und/oder endoskopische Verfahren zum Einsatz kommen. Aufgrund der derzeit limitierten Therapiemöglichkeiten besteht daher ein hoher Forschungsbedarf an wirksamen medikamentösen Therapien.
Fabian Sandner, Nürnberg
Familiäres
Chylomikronämie-Syndrom: Uniklinik Köln stellt den ersten Patienten auf Olezarsen ein
Am 16. Dezember 2025 wurde an der Uniklinik Köln der erste Patient mit Familiärem Chylomikronämie-Syndrom (FCS) auf den neuen ApoC3-Inhibitor Olezarsen (Tryngolza®) eingestellt. Damit hat 2 Monate nach der Zulassung in der Europäischen Union der klinische Einsatz der Therapie in einem der führenden lipidologischen Zentren in Deutschland begonnen. Im Rahmen eines Expertengesprächs hob Prof. Dr. Ioanna Gouni-Berthold, Leiterin der Lipidambulanz und der Studienambulanz für Fettstoffwechselstörungen an der Uniklinik Köln, die Bedeutung der neuen Therapieoption hervor, die bereits in der zulassungsrelevanten Phase-III-Studie die Triglyzeridwerte um 59,4 % senken und 88 % der Pankreatitis-Ereignisse verhindern konnte. „Triglyzeride werden in der klinischen Praxis oft unterschätzt“, betonte Gouni-Berthold und ergänzte: Es geht nicht nur um das
kardiovaskuläre Risiko. Mit steigenden Triglyzeridwerten nimmt vor allem das Risiko für eine akute Pankreatitis deutlich zu.“ Ab Triglyzeridwerten von 885 mg/dl (10 mmol/l) spricht man von einer schweren Hypertriglyzeridämie (HTG).
FCS – eine seltene und schwerwiegende genetische Fettstoffwechselerkrankung
Das FCS ist durch dauerhaft stark erhöhte Triglyzeridwerte gekennzeichnet. Es beginnt bereits im Kindesalter, wird jedoch häufig erst im jüngeren Erwachsenenalter diagnostiziert. Verursacht wird das FCS überwiegend durch eine beeinträchtigte Funktion des Enzyms Lipoproteinlipase (LPL), das für den Abbau von Triglyzeriden aus dem Blut verantwortlich ist. Triglyzeride werden nach der Aufnahme aus dem Darm im Blut in Chylomikronen transportiert. Aufgrund der eingeschränkten LPL-Funktion können Menschen mit FCS Triglyzeride und damit auch Chylomikronen nicht effektiv aus dem Blut abbauen. Die Folge
ist eine schwere Hypertriglyzeridämie und Chylomikronämie.
Akute und rezidivierende Pankreatitis als schwerwiegendste Komplikation
Neben Konzentrationsproblemen, Xanthomen, Hepatosplenomegalie und wiederkehrenden, zum Teil schweren Abdominalschmerzen zählt die potenziell lebensbedrohliche akute Pankreatitis zu den schwerwiegendsten Folgen des FCS. Das Risiko für eine akute Pankreatitis steigt exponentiell mit der Höhe der Triglyzeridwerte an: Ab ≥885 mg/dL (10 mmol/l) gilt es als hoch, ab ≥1000 mg/dL (11,2 mmol/l) als extrem hoch. Besonders hoch ist das Risiko bei genetisch bestätigtem FCS: Diese Patienten haben ein 360-fach erhöhtes Risiko für eine akute Pankreatitis im Vergleich zu normolipämischen Patienten. FCS-bedingte Pankreatitiden verlaufen zudem oft schwer und sind mit einem höheren Risiko für Organversagen, Pankreasnekrose, intensivmedizinische Behandlung und Mortalität verbunden.
Abbildung 1: Aufbau von Volanesorsen und Olezarsen im Vergleich (© Sobi).
Olezarsen (Tryngolza® 80 mg Injektionslösung) ist ein triantennär N-Acetylgalactosamin-konjugiertes Antisense-Oligonukleotid, das selektiv an die mRNA von Apolipoprotein C3 bindet und deren Abbau über die RNase-H1 vermittelt. Dadurch sinken die ApoC3Proteinspiegel und in der Folge die Triglyzeridwerte im Plasma. Zusätzlich kommt es zu Veränderungen weiterer lipidbezogener Parameter wie Chylomikron-Triglyzeriden, ApoB-48, Gesamtcholesterin und Nicht-HDL-Cholesterin sowie zu einem Anstieg des HDL-Cholesterins.
Olezarsen ist zugelassen zur Behandlung erwachsener Patienten mit genetisch bestätigtem FCS, ergänzend zu einer Diät und kann mit anderen lipidsenkenden Arzneimitteln, etwa Statinen und Fibraten, kombiniert werden. Die empfohlene Dosierung beträgt 80 mg subkutan einmal monatlich.
Primäres Therapieziel ist die Verhinderung von Pankreatitiden
Um dieses Ziel zu erreichen, werden strikte, stark fettreduzierte Diäten eingesetzt, die jedoch nur begrenzt wirksam sind. Klassische lipidsenkende Interventionen sind zur Behandlung des FCS nicht oder nur bedingt geeignet, da sie ihren Effekt über einen LPL-vermittelten Abbau der Triglyzeride ausüben, der bei FCS jedoch beeinträchtigt ist. Der seit 2021 in Deutschland verfügbare ApoC3-Hemmer Volanesorsen (Waylivra®) zeigt zwar eine gute Wirksamkeit, ist aber aufgrund von immunologisch bedingten Kontraindikationen und/ oder (häufigen) Nebenwirkungen wie z.B. Thrombozytopenien nicht für alle FCS-Patienten geeignet. Mit Tryngolza® (Olezarsen 80 mg) steht nun in Deutschland ein ApoC3-Hemmer der nächsten Generation zur Verfügung, der durch sein verbessertes Sicherheitsprofil
diese therapeutische Lücke schließen kann.
Ein gezielt gegen ApoC3 gerichtetes AntisenseOligonukleotid mit verbessertem Sicherheitsprofil
Apolipoprotein C3 spielt eine Schlüsselrolle im Triglyzeridstoffwechsel. Es wird hauptsächlich in der Leber gebildet und hemmt über einen LPL-abhängigen wie auch LPL-unabhängigen Weg den Abbau der Triglyzeride. Wird die Bildung von ApoC3 gehemmt, wird diese Hemmung des Triglyzeridabbaus aufgehoben und die Triglyzeride können wieder verstärkt aus dem Blut abgebaut werden. Genau hier setzt die Therapie mit Olezarsen an. Es ist wie sein Vorgängerprodukt Volanesorsen ein Antisense-Oligonukleotid (ASO), das gegen ApoC3 gerichtet ist. Zusätzlich ist es aber mit einem GalNAc3-Konjugat modifiziert (Abb. 1). Dadurch wird es zielgerichtet in
die Leber aufgenommen, kann geringer dosiert und seltener verabreicht werden. Bei vergleichbarer Wirksamkeit zeigt es gegenüber Volanesorsen durch die verminderte systemische Exposition ein verbessertes Sicherheitsprofil. Die Vorteile im Therapiemanagement sind deutlich: Es ist kein aufwendiges Labormonitoring mehr erforderlich und die Handhabung durch monatliche Applikation mittels Fertigpen ist einfacher. „Bei unserer Patientin waren unter Volanesorsen vor allem die grippeähnlichen Symptome und die regelmäßigen Blutentnahmen zur Kontrolle der Thrombozyten ein Problem. Der Wegfall bedeutet für die Patientin und uns eine erhebliche Erleichterung im Alltag“, berichtete Gouni-Berthold.
Überzeugende Studiendaten aus der Phase-III-Studie BALANCE
In der randomisierten, placebokontrollierten BALANCE-Studie mit 66 Patienten senkte Olezarsen 80 mg nach 12 Monaten Behandlung signifikant die Nüchtern-Triglyzeride im Vergleich zu Placebo um 59,4 %. Die Anzahl akuter Pankreatitis-Ereignisse wurde um 88 % reduziert. Dabei zeichnete sich Olezarsen durch ein günstiges Sicherheits- und Verträglichkeitsprofil auf und es traten weniger unerwünschte Ereignisse auf als unter Placebo. Arzneimittelbedingte, schwerwiegende unerwünschte Ereignisse wurden unter der Behandlung mit Olezarsen nicht beobachtet.
Brigitte Söllner, Erlangen
Paradigmenwechsel in der Hämophilie A:
Emicizumab verbessert
Gelenkgesundheit –NXT007 ebnet Weg zur normalisierten Hämostase
Auf der Jahrestagung der American Society of Hematology (ASH) 2025 präsentierte Roche neue Daten, die den Fokus in der Behandlung von Hämophilie A (HA) verschieben: weg von der reinen Blutungsrate hin zu funktionellen Endpunkten wie Gelenkgesundheit und physische Belastbarkeit. Zudem bietet der vielversprechende Pipeline-Kandidat NXT007 einen spannenden Ausblick auf die nächste Generation therapeutischer Innovationen in der HA-Therapie.
Verbesserte Gelenkgesundheit führt zu Steigerung der körperlichen Aktivität
Dank der Fortschritte in der Hämophilie A-Behandlung sind Blutungen heute deutlich seltener geworden. Daher verlagert sich der Schwerpunkt zunehmend auf die langfristige Gelenkgesundheit, da sich der Gelenkstatus auch bei einer Faktor-VIII-Prophylaxe über die Zeit verschlechtern kann. Vor diesem Hintergrund ermöglicht es die effektive Blutungsprophylaxe mit Emicizumab (Helimbra®) den Patienten, z.B. mehr sportliche Aktivitäten in ihren Alltag zu integrieren – und das bei minimalem Blutungsrisiko. Dies bestätigen neueste Post-Marketing-Daten der Studie BEYOND ABR und ergänzen damit die klinische und Real-World-Evidenz. Die multizentrische, offene Phase-IV-Studie untersuchte auch die Gelenkge-
sundheit bei 13- bis 69-jährigen Patienten mit mittelschwerer oder schwerer HA (ohne Faktor-VIIIInhibitoren) nach dem Wechsel von einer Faktor-VIII- auf eine Emicizumab-Prophylaxe. Die zweite Interimsanalyse zeigte im ersten Jahr nach der Umstellung auf Emicizumab niedrige Blutungsraten und eine Verbesserung der Gelenkgesundheit. Der mittlere Hemophilia Joint Health Score (HJHS) verbesserte sich innerhalb des Beobachtungszeitraums bei der Mehrzahl der Patienten oder blieb stabil. Diese Ergebnisse sind konsistent mit den Beobachtungen aus der Studie HAVEN 3. In diesem Zusammenhang konnte eine Verschiebung hin zu höheren Aktivitätsleveln festgestellt werden. Gemessen am International Physical Activity Questionnaire (IPAQ) stieg der Anteil an Patienten mit hoher Aktivität von 44,2 % auf 50,0 %. Der erhöhte Aktivitätslevel steht in Übereinstimmung mit den Beobachtungen aus der Studie TSUBASA und ermöglicht den Patienten eine verbesserte Lebensqualität, da regelmäßige Bewegung und körperliche Aktivität eine essentielle Grundlage für ein physisch und mental gesundes Leben sind.
NXT007: Potenzial zur Normalisierung der Hämostase
Einen Ausblick auf die nächste Generation der HämophilieA-Therapien geben die Phase-I/ II-Studiendaten zu NXT007. Der bispezifische Antikörper, der von Chugai entwickelt wurde und gemeinsam mit Roche erforscht wird, ist eine Weiterentwicklung von Emicizumab. Erste Studien
zeigen sein Potenzial, die Thrombingenerierung zu steigern und die Hämostase auf das Niveau einer gesunden Person zu bringen. Die auf der ASH-Jahrestagung 2025 vorgestellten Phase-I/II-Daten zu NXT007 sind richtungsweisend für die Entwicklung in diesem Jahr, in dem NTX007 mit dem Start der Phase-III-Studien weiter in den Fokus rücken wird. Die offene, nicht randomisierte, globale multiple-ascending-dose (MAD) Studie mit NXT007 lieferte wichtige Erkenntnisse zur Sicherheit, Verträglichkeit und Pharmakokinetik des Antikörpers. Der Wirkstoff wurde in allen Dosiskohorten gut vertragen und zeigte ein günstiges Sicherheitsprofil. Die annualisierten Blutungsraten (ABRs) für behandelte Blutungen waren pro Kohorte insgesamt niedrig: Die mittlere jährliche Blutungsrate (ABR) sank von 1,78 auf 0,62, wobei der Großteil der Studienpopulation (86,4 %) unter der Erhaltungstherapie vollständig blutungsfrei blieb. Diese Daten sind weitgehend konsistent mit der zuvor präsentierten Primäranalyse von Teil B der Phase-I/II NXTAGE-Studie. Weiterführende Studien sollen das Potenzial von NXT007 weiter evaluieren und nachweisen.
Neben der gesteigerten Wirksamkeit zielt das klinische Entwicklungsprogramm von Roche und Chugai auch darauf ab, den Anwendungskomfort für die Patienten durch optimierte Verabreichungsmöglichkeiten weiter zu verbessern. So befindet sich derzeit auch ein Autoinjektor für Emicizumab in der Entwicklung. Elisabeth Wilhelmi, München

NIGER: Edriss Haruna (2) ist akut mangelernährt. Unsere Ärztin Dr. Faïza Ouedraogo behandelt ihn, daneben seine Mutter Harira Mohamed. © Oliver Barth
Mit 38 Euro kann ärzte ohne grenzen zwei akut mangelernährte Kinder mit therapeutischer Spezialnahrung versorgen, bis sie wieder bei Kräften sind.
Spendenkonto: Bank für Sozialwirtschaft
IBAN: DE72 3702 0500 0009 7097 00
BIC: BFSWDE33XXX www.aerzte-ohne-grenzen.de/spenden
Herausgeber:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
Univ.-Prof. Dr. med. Hermann Eichstädt, Leiter Bereich Kardiologie RZP Potsdam und Geschäftsführer BBGK e.V. Berlin
Konstanzer Straße 61 10707 Berlin
Wissenschaftlicher Beirat:
Prof. Dr. M. Alexander, Infektiologie, Berlin
Prof. Dr. L. Beck, Gynäkologie, Düsseldorf
Prof. Dr. Berndt, Innere Medizin, Berlin
Prof. Dr. H.-K. Breddin, Innere Medizin, Frankfurt/Main
Prof. Dr. K. M. Einhäupl, Neurologie, Berlin
Prof. Dr. E. Erdmann, Kardiologie, Köln
Prof. Dr. Dr. med. E. Ernst, University of Exeter, UK
Prof. Dr. K. Falke, Anästhesiologie, Berlin
Prof. Dr. K. Federlin, Innere Medizin, Gießen
Prof. Dr. E. Gerlach, Physiologie, München
Prof. Dr. H. Helge, Kinderheilkunde, Berlin
Prof. Dr. R. Herrmann, Onkologie, Basel
Prof. Dr. W. Jonat, Gynäkologie, Hamburg
Prof. Dr. H. Kewitz, Klin. Pharmakol. Berlin
Titelbild: Quelle Johnson & Johnson
Prof. Dr. B. Lemmer, Pharmakologie, Mannheim/Heidelberg
Prof. Dr. med. R. Lorenz, Neurochirurgie, Frankfurt
Prof Dr. J. Mann, Nephrologie, München
Dr. med. Veselin Mitrovic, Kardiologie, Klinische Pharmakologie, Bad Nauheim
Prof. Dr. R. Nagel, Urologie, Berlin
Prof. Dr. E.-A. Noack, Pharmakologie, Düsseldorf
Prof. Dr. P. Ostendorf, Hämatologie, Hamburg
Prof. Dr. Th. Philipp, Innere Medizin, Essen
Priv.-Doz. Dr. med. B. Richter, Ernährung – Stoffwechsel, Düsseldorf
Prof. Dr. H. Rieger, Angiologie, Aachen
Prof. Dr. H. Roskamm, Kardiologie, Bad Krozingen
Prof. Dr. E. Rüther, Psychiatrie, Göttingen
Prof. Dr. med. A. Schrey, Pharmakologie, Düsseldorf
Dr. Dr. med. C. Sieger, Gesundheitspolitik u. Gesundheitsökonomie, München
Prof. Dr. E. Standl, Innere Medizin, München
Prof. Dr. W. T. Ulmer, Pulmologie, Bochum
Schriftleitung:
Prof. Dr. med. Karl-Ludwig Resch, Deutsches Institut für Gesundheitsforschung gGmbH, Ossecker Str. 172, 95030 Hof
E-Mail: info@d-i-g.org
E-Mail persönlich: k.l.resch@d-i-g.org
Die Zeitschrift erscheint 6 mal im Jahr; Jahresabonnement € 66,00 inkl. MwSt. zzgl. Versandspesen. Einzelheft € 11,00 inkl. MwSt. zzgl. Versandspesen. Studenten-Abo zum halben Preis. Der Abonnementpreis ist im Voraus zahlbar. Stornierungen sind bis 6 Wochen vor Ablauf eines Kalenderjahres möglich. Abonnementbestellungen direkt beim Verlag.
Geschäftsführerin: Sibylle Michna Anschrift wie Verlag
Chefredaktion: Brigitte Söllner (verantwortlich) Anschrift wie Verlag
Herstellung/Layout: HGS5 GmbH Schwabacherstr. 117 90763 Fürth
Werbung, Beratung, Verkauf: Sibylle Michna Anschrift wie Verlag
Die Annahme von Werbeanzeigen impliziert nicht die Empfehlung durch die Zeitschrift; die in den Beiträgen zum Ausdruck gebrachten Meinungen und Auffassungen drücken nicht unbedingt die der Herausgeber, des wissenschaftlichen Beirates oder des Verlages aus. Der Verlag behält sich alle Rechte, insbesondere das Recht der Vervielfältigung jeglicher Art, sowie die Übersetzung vor. Nachdruck, auch auszugsweise, nur mit Genehmigung des Verlages.
Erfüllungsort: Puschendorf Gerichtsstand: Fürth
Fälle höherer Gewalt, Streik, Aussperrung und dergleichen entbinden den Verlag von der Verpflichtung auf Erfüllung von Aufträgen und Leistungen von Schadensersatz.
Anmerkung der Redaktion: Zur besseren Lesbarkeit werden im Journal für Pharmakologie und Therapie personenbezogene Bezeichnungen, die sich auf das männliche oder weibliche Geschlecht beziehen, grundsätzlich nur in der männlichen Form verwendet. Damit wird keine Diskriminierung des Geschlechts ausgedrückt.
Satz:
HGS5 GmbH, Schwabacherstr. 117 90763 Fürth
Druck und Verarbeitung: DRUCK_INFORM_W.R.
Roland Welker
Austraße 7, 96114 Hirschaid
Verlag PERFUSION GmbH
Storchenweg 20 90617 Puschendorf
Telefon: 09101/990 11 10
Fax: 09101/990 11 19
www.Verlag-Perfusion.de
E-Mail: perfusion@t-online.de
Langzeitdaten aus Klinik und Praxis4,5 Langanhaltend

Behandeln Sie das Sichtbare. Verhindern Sie das Unsichtbare.
auf Haut & Gelenke1–3 Wirkstark
günstiges
Sicherheitsprofil für Ihre Patient*innen6,7 Bewährt
a STEPIn: Randomisierte Open-Label-Studie zum Vergleich der Standardtherapie (Schmalband-UVB) mit Cosentyx® über einen Behandlungszeitraum von 52 Wochen bei Patient*innen mit neu aufgetretener (≤ 12 Monate) mittelschwerer bis schwerer Plaque-Psoriasis. 9 von 10 Patient*innen erreichten mit Cosentyx® 300 mg nach 1 Jahr ein PASI90-Ansprechen, fast 7 von 10 ein PASI100-Ansprechen.1 b FUTURE 5: In die randomisierte, placebokontrollierte Phase-III-Studie waren TNFi-naive Patient*innen mit aktiver PsA und solche, die unzureichend auf eine Therapie mit TNFi angesprochen haben, eingeschlossen. Cosentyx® konnte ab Woche 52 von 150 mg auf 300 mg erhöht werden, wenn aktive Krankheitssymptome in einer ärztlichen Beurteilung beobachtet wurden. Unter Cosentyx® 300 mg waren in Woche 104 89,5 % der PsA-Patient*innen ohne röntgenologische Progression. In der 150-mg-Dosierung waren es 82,3 %.2 c IVEPSA: Einarmige, prospektive, explorative Open-Label-Studie bei Psoriasis-Patient*innen mit Beteiligung der Kopfhaut oder Nägel oder einem PASI > 6 sowie entzündlichen oder erosiven Veränderungen im MRT oder CT. Nach 24 Wochen Behandlung mit Cosentyx® 300 mg konnte eine Reduktion der subklinischen Entzündung festgestellt werden.3 CT Computertomographie. IVEPSA Interception in very early PsA. MRT Magnetresonanztomographie. PASI Psoriasis Area and Severity Index. PsA Psoriasis-Arthritis. TNFi Tumornekrosefaktor-Inhibitor. UVB Ultraviolett B. 1. Iversen L, et al. J Eur Acad Dermatol Venereol. 2023;37(5):1004-1016. 2. Mease PJ, et al. RMD Open. 2021;7(2):e001600. 3. Kampylafka E, et al. Arthritis Res Ther. 2019;21(1):178. 4. Bissonnette R, et al. J Eur Acad Dermatol Venereol. 2018;32(9):1507-1514. 5. Augustin M, et al. EADV 2024. Poster 3355. 6. Fachinformation Cosentyx. 7. Sun R, et al. Dermatol Ther. 2024;14:729-743. Cosentyx® ist angezeigt für die Behandlung von Erwachsenen sowie Kindern und Jugendlichen ab 6 Jahren mit mittelschwerer bis schwerer Plaque-Psoriasis,die für eine systemische Therapie in Frage kommen. Cosentyx®, allein oder in Kombination mit Methotrexat (MTX), ist angezeigt für die Behandlung erwachsener Patient*innen mit aktiver Psoriasis-Arthritis, wenn das Ansprechen auf eine vorhergehende Therapie mit krankheitsmodifizierenden Antirheumatika (DMARD) unzureichend gewesen ist. Cosentyx® 75 mg Injektionslösung in einer Fertigspritze, Cosentyx® 150 mg Injektionslösung in einer Fertigspritze, Cosentyx® 150 mg Injektionslösung in einem Fertigpen, Cosentyx® 300 mg Injektionslösung in einer Fertigspritze, Cosentyx® 300 mg Injektionslösung in einem Fertigpen. Wirkstoff: Secukinumab (in Ovarialzellen d. chines. Hamsters [CHO-Zellen] produzierter, gg. Interleukin-17A gerichteter, rekombinanter, vollständig humaner monoklonaler Antikörper d. IgG1/κ-Klasse). Zus.-setz.: Arzneil. wirks. Bestandt.: 1 Fertigspritze enthält 75 mg Secukinumab in 0,5 ml bzw. 1 Fertigspritze/Fertigpen enthält 150 mg Secukinumab in 1 ml bzw. 300 mg Secukinumab in 2 ml. Sonst. Bestandt.: Trehalose-Dihydrat, Histidin, Histidinhydrochlorid-Monohydrat, Methionin, Polysorbat 80, Wasser f. Inj.-zwecke. Anwend.: Behandl. v. Kindern u. Jugendl. ab 6 J. mit mittelschwerer bis schwerer Plaque-Psoriasis, d. für eine system. Therapie in Frage kommen. Behandl. v. Kindern u. Jugendl. ab 6 J. mit Enthesitis-assoziierter Arthritis od. juveniler Psoriasis-Arthritis, allein od. in Kombination mit Methotrexat (MTX), wenn Erkrankung unzureich. auf eine konventionelle Therapie angesprochen hat od. d. diese nicht vertragen. 150/300 mg Injektionslösung zusätzl.: Behandl. erw. Pat. mit mittelschwerer bis schwerer Plaque-Psoriasis, d. für eine system. Therapie in Frage kommen. Behandl. erw. Pat. mit mittelschwerer bis schwerer aktiver Hidradenitis suppurativa (Acne inversa), d. auf eine konventionelle system. HS-Therapie unzureichend angesprochen haben. Behandl. erw. Pat. mit aktiver Psoriasis-Arthritis, allein od. in Kombination mit MTX, wenn d. Ansprechen auf eine vorhergeh. Therapie mit krankheitsmodifizierenden Antirheumatika (DMARD) unzureich. gewesen ist. Behandl. erw. Pat. mit aktiver ankylosierender Spondylitis, d. auf eine konventionelle Therapie unzureich. angesprochen haben. Behandl. erw. Pat. mit aktiver nichtröntgenolog. axialer Spondyloarthritis mit objektiven Anzeichen d. Entzündung, angez. durch erhöhtes C-reaktives Protein (CRP) u./od. Nachweis durch Magnetresonanztomographie (MRT), d. unzureich. auf nichtsteroid. Antirheumatika (NSAR) angesprochen haben. Gegenanz.: Überempfindlichkeit gg. d. Wirkstoff od. einen d. sonst. Bestandt. Klinisch relevante, aktive Infekt. (z. B. aktive Tuberkulose). Nebenw.: Sehr häufig: Infekt. d. oberen Atemwege. Häufig: Oraler Herpes. Kopfschmerzen. Rhinorrhö. Diarrhö, Übelkeit. Ekzem. Ermüdung. Gelegentl.: Orale Candidose, Otitis externa, Infekt. d. unteren Atemwege, Tinea pedis. Neutropenie. Konjunktivitis. Entzündl. Darmerkrankungen. Dyshidrot. Ekzem. Urtikaria. Selten: Anaphylakt. Reakt., Angioödem. Exfoliative Dermatitis, Hypersensitivitätsvaskulitis. Häufigkeit nicht bekannt: Mukokutane Candidose (einschl. ösophageale Candidose). Pyoderma gangraenosum. Verschreibungspflichtig. Weit. Angaben: S. Fachinformationen. Stand: Februar 2025 (MS 04/25.25). Novartis Pharma GmbH, Sophie-Germain-Str. 10, 90443 Nürnberg. Tel.: (09 11) 273-0. www.novartis.de