Journal for Clinical Studies – ISSN 1758-5678 is published quarterly by Senglobal Ltd.
4 FOREWORD
WATCH PAGES
6 Mental Health Applications of Generative Artificial Intelligence
One of the newer areas in which the US Food and Drug Administration (FDA) is exploring its regulatory role is the use of artificial intelligence (AI) in digital mental health medical devices. There is recognition that mental illnesses are common around the world. Deborah Komlos of Clarivate discusses how the FDA seeks to leverage feedback from the DHAC to help the agency foster innovation in the field of genAI-enabled digital mental health medical devices while ensuring the safety of patients.
MARKET REPORTS
8 How Global Contact Centre Support Enhances Clinical Trial Success
This article explores how global contact centres enable a more reliable and patient-centric clinical trial experience through consistent and compliant communication, safety reporting and delivering continuity across global, multi-site studies. Rajul Jain and Valerie Huh of ProPharma discuss how investing in a well-structured global contact centre enables sponsors to deliver a more resilient, compliant and patient-centric trial experience, one that supports scientific rigor while honouring the needs and expectations of participants worldwide.
12 Optimising Clinical Trials for the Era of Self-Injectable Combination Products
Self administered injectable drug–device combination (DDC) products are entering a phase of accelerated therapeutic innovation. Demographic change, chronic disease trends and the push for patient centric care are converging to make self administration a practical imperative rather than a futuristic aspiration. Alexander Limprecht of PCI discusses how well designed DDCs can reduce injection related anxiety, pain and procedural complexity, contributing to a more positive trial experience and sustained engagement. For sponsors, early integration of combination products can de risk post approval use, facilitate evidence generation for regulators and payers, reduce cost.
APPLICATION
NOTE
14 De-Risking FIH: Integrated Strategies for Rapid Proof-of-Concept
The opinions and views expressed by the authors in this journal are not necessarily those of the Editor or the Publisher. Please note that although care is taken in the preparation of this publication, the Editor and the Publisher are not responsible for opinions, views, and inaccuracies in the articles. Great care is taken concerning artwork supplied, but the Publisher cannot be held responsible for any loss or damage incurred. This publication is protected by copyright.
Volume 18 Issue 1 Spring 2026, Senglobal Ltd.
www.journalforclinicalstudies.com
There is intense pressure on drug developers to progress studies faster into the clinical development timeline with fewer financial resources. Dr. Andreas Reichl and Dr. Kevin Schaab of Quotient discuss how their Translational Pharmaceutics® approach is proven to accelerate development programmes by leveraging a purpose-built infrastructure to develop formulations, provide GMP or compounded drug products to deliver quality clinical data. This approach has evolved to deliver integrated programmes under one organisation, steered through a single project management point of contact.
REGULATORY AFFAIRS
18 From Start-up to Sustainability: The New Reality of Research Site Operations
Research site challenges are no longer episodic disruptions they are structural conditions. Clinical trial complexity, staffing shortages,
Contents
funding volatility, technology burden and rising participant expectations interact in ways that amplify risk and constrain capacity. Sandy Smith of WCG discusses how the future of clinical research depends on the sustainability of its sites. It is foundational to scientific progress, to equity in research participation and to the delivery of therapies that improve and save lives.
22 Beyond the IDMC:
The Value of Safety Review Committees
Safety Review Committees (SRCs) represent a critical component of early-phase clinical research governance. As the landscape of drug development evolves toward accelerated, adaptive and first-in-human (FIH) designs, the need for scientifically rigorous safety oversight becomes increasingly evident. Maxim Kosov, Bethany Kloss and Jennifer Bradley of PSI discuss that by early-phase trials increasingly provide pivotal evidence for regulatory approvals, the role of SRCs will continue to expand in scope and importance.
THERAPEUTICS
26 Patient-Derived Tumour Organoids for Precision Screening: Operation GxP-Level Quality for Translation Reliability
Patient-derived organoids have emerged as an alternative that replicates the three-dimensional structure, genetic diversity and heterogeneity of human tumours. However, Seahee Kim of Samsung Biologics discusses how the translational utility of these organoids depends on two inseparable pillars: biological fidelity and operational rigor. Biological fidelity ensures that in vitro responses reflect patient outcomes, while operational rigor guarantees that results are reproducible and scalable across studies and client programmes.
28 Recentring Sites:
The Human Factors Slowing Study Startup and How to Resolve Them
The clinical research industry has rightly invested in understanding and improving participant experiences. These efforts have yielded meaningful benefits for participants. However, clinical trials continue to stall at the point where participant involvement actually begins. Brian Mallon of ICON discusses how human-centred approaches can accelerate site activation by reducing friction at the point where trials often stall and by giving sites better conditions for recruitment and retention. Recognising site needs, improving communication and aligning expectations create conditions where participant-centric strategies can succeed. In doing so, the industry gives itself a more reliable route to better participant outcomes.
CLINICAL TRIAL MANAGEMENT
30 Navigating Complexity in Master Protocols with Key Operational Partners
Master Protocols have the capacity for great advantages and efficiencies in drug development over traditional trial protocols but can be challenging for sponsors due to their inherent complexity. Jennifer Ross and Cheryl Fitzer-Attas of Almac Clinical Technologies discuss how Master Protocols offer significant advantages in clinical trial efficiency and patient benefit but involve inherent complexity requiring expert operational support.
36 Excel in Data Management During Clinical Trial Handoffs
Clinical trial clients often face the complex task of a contract research organisation (CRO) transition, moving critical trial functions from one
CRO to a functional service provider (FSP) organisation. Melanie Dyer and Kristine Smith of Worldwide Flex Clinical Trials discuss that by focusing on proactive planning, collaborative culture and rigorous oversight, a well-managed transition can solve the challenges of vendor changeovers and deliver real benefits.
38 Measuring What Matters: Designing a Future-Fit FSP Model for Modern Drug Development
The Functional Service Provider model has grown from a tactical staffing mechanism into a strategic outsourcing framework that underpins modern clinical development. Yet, the way many organisations measure FSP performance has not kept pace. Sarah Tucker of Phastar discusses how the future of FSP is not about headcount expansion. It is about intelligent ecosystem design that not only meets today’s needs but is designed to evolve with the demands of the future.
TECHNOLOGY
40 The Silent Document Revolution Unblocking Clinical Trials
Clinical trials run on documents and data. When these documents are slow, inconsistent, or misaligned, the entire study slows with them. Fareed Melhem of Veridix AI discusses how new authoring agents embed purpose-built AI trained exclusively on clinical-trial documents and vetted reference materials and validated by medical writers and biotechnicians. They understand protocols, SAPs, CSRs and myriad more document types and the structured relationships that connect them. These quieter revolutions are already moving the industry forward.
42 Next-Generation Risk-Based Monitoring: The Role of AI in Clinical Trial Oversight
AI-augmented Risk-Based Monitoring (RBM) represents a transformative shift in clinical trial monitoring and oversight, moving beyond reactive, threshold-based approaches to predictive, adaptive and continuously learning systems. Ashok Ghone of MedInventas discusses how AI does not replace clinical judgment; rather, it augments human expertise by automating routine monitoring tasks and providing actionable insights, allowing study teams to focus on high-value decision-making and strategic oversight.
We begin our year with our spring edition of JCS. In this edition, our contributors explore how the industry is striving to make clinical trials a more reliable and patient-centric experience. We delve into how well-designed DDCs can help reduce injection-related anxiety, pain and procedural complexity to create a more positive trial experience, thus sustaining engagement and retention. This issue also concentrates on the rise of Artificial Intelligence and how it will help orchestrate a smoother and more efficient process during clinical trials.
The transition from preclinical to clinical testing is a pivotal moment in drug development. Dr. Andreas Reichl and Dr. Kevin Schaab of Quotient delve into how Biotech’s funding challenges and current downward trend in investment necessitate speedy, de-risked early-stage development. This article highlights how the Translational Pharmaceutics® approach is proven to accelerate development programmes by leveraging a purposebuilt infrastructure to develop formulations, provide GMP or compounded drug products to deliver quality clinical data. This approach has evolved to deliver integrated programmes under one organisation, steered through a single project management point of contact. This is exemplified by their unique platform, Translational Pharmaceutics®, which simplifies programme design and enhances decision-making on every project while also significantly reducing clients’ R&D spend.
Melanie Dyer and Kristine Smith of Worldwide Flex discuss how clinical trial clients often face the complex task of a contract research organisation (CRO) transition, moving critical trial functions from one CRO to a functional service provider (FSP) organisation. They delve into how there are as many ways to navigate these changes as there are providers, but some best practices help to drive excellence in handoffs of data management, ensuring trials stay on track. In this study Melanie and Kristine explore how guidance supports effective transition strategies that safeguard quality and timelines. They share that by focusing on proactive planning, collaborative culture, and rigorous oversight, a well-managed transition can solve the challenges of vendor changeovers and deliver real benefits, like sustained data integrity during handoff and uninterrupted progress in clinical trial data management. This can result in a smoother operation that mitigates risk and sets your team up for long-term success.
Self administered injectable drug-device combination (DDC) products are entering a phase of accelerated therapeutic innovation. Demographic change, chronic disease trends and the push for patient centric care are converging to make self administration a practical imperative rather than a futuristic aspiration. Alexander Limprecht of PCI delves into how self administered injectable DDCs have become a keystone technology for upgrading drug delivery, improving adherence and ultimately optimising clinical outcomes. For sponsors, the logical extension of this trend is to incorporate self injectable DDCs early, at the clinical trial stage rather than only at commercialisation.
Melissa Cavner, Editor
JCS – Editorial Advisory Board
• Ashok K. Ghone, PhD, VP, Global Services MakroCare, USA
• Bakhyt Sarymsakova – Head of Department of International Cooperation, National Research Center of MCH, Astana, Kazakhstan
• Catherine Lund, Vice Chairman, OnQ Consulting
• Cellia K. Habita, President & CEO, Arianne Corporation
• Chris Tait, Life Science Account Manager, CHUBB Insurance Company of Europe
• Deborah A. Komlos, Principal STEM Content Analyst, Clarivate
• Elizabeth Moench, President and CEO of Bioclinica – Patient Recruitment & Retention
• Francis Crawley, Executive Director of the Good Clinical Practice Alliance – Europe (GCPA) and a World Health Organisation (WHO) Expert in ethics
• Georg Mathis, Founder and Managing Director, Appletree AG
• Hermann Schulz, MD, Founder, PresseKontext
• Jeffrey W. Sherman, Chief Medical Officer and Senior Vice President, IDM Pharma.
• Jim James DeSantihas, Chief Executive Officer, PharmaVigilant
• Mark Goldberg, Chief Operating Officer, PAREXEL International Corporation
• Maha Al-Farhan, Chair of the GCC Chapter of the ACRP
• Rick Turner, Senior Scientific Director, Quintiles Cardiac Safety Services & Affiliate Clinical Associate Professor, University of Florida College of Pharmacy
• Robert Reekie, Snr. Executive Vice President Operations, Europe, AsiaPacific at PharmaNet Development Group
• Stanley Tam, General Manager, Eurofins MEDINET (Singapore, Shanghai)
• Stefan Astrom, Founder and CEO of Astrom Research International HB
• Steve Heath, Head of EMEA – Medidata Solutions, Inc
Mental Health Applications of Generative Artificial Intelligence
One of the newer areas in which the US Food and Drug Administration (FDA) is exploring its regulatory role is the use of artificial intelligence (AI) in digital mental health medical devices.
There is recognition that mental illnesses are common around the world. As noted in a 2024 report from the World Health Organisation (WHO), World mental health today: Latest data, more than 1 billion people worldwide are living with a mental disorder.1 The report notes that the prevalence of different mental disorders varies by sex, with females overall more affected. Anxiety disorders and depressive disorders are the most common conditions in both sexes.
Citing data from the National Survey on Drug Use and Health (NSDUH) by the Substance Abuse and Mental Health Services Administration (SAMHSA), the National Institute of Mental Health of the National Institutes of Health (NIH) notes that in 2022, an estimated 59.3 million adults aged 18 or older in the US were diagnosed with any mental illness (AMI).2 This number represented 23.1% of all US adults, more than 1 in 5. Out of these nearly 60 million adults, 50.6% received mental health treatment in the past year. The NSDUH defines mental health treatment as having received inpatient treatment/counselling or outpatient treatment/counselling or having used prescription medication to help with mental health.
Issued in July 2025, the NSDUH’s latest annual report provides indicators of mental health in the US based on data from 2021 to 2024.3
As in the 2022 survey, adults aged 18 or older were classified as having AMI if they had any mental, behavioural, or emotional disorder in the past year of sufficient duration to meet criteria from the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV), excluding developmental disorders and substance use disorders. Across the assessed population, the percentage of adults with AMI in the past year did not change from 2021 to 2024. In 2024, 23.4% of adults aged 18 or older – representing 61.5 million people – had AMI in the past year. Rates within each age group also remained stable over this period. Among young adults aged 18 to 25, approximately one-third (33.2%, or 11.6 million people) had AMI in the past year.
Medical Devices for Mental Health
Given this substantial and persistent prevalence, the FDA convened its second-ever meeting of the Digital Health Advisory Committee (DHAC) in early November 2025 to examine issues related to the use of generative AI (genAI) – enabled digital mental health medical devices. The committee discussed potential benefits and risks to health, possible risk-mitigation strategies, premarket evidence expectations and approaches to postmarket monitoring.
The agency explained in the preface to the panel discussion questions that, alongside the increasing accessibility of genAI products for general use, the development and demand for a new type of digital mental health medical device are also rising.4 These "AI therapists," and other AI-based medical devices aim to offer a broad range of mental health therapies and interactions with therapist or healthcare provider (HCP) – like chatbots, some of which may even be diagnostic. Because these chatbots can interact with users in personalised ways – with or without HCP oversight – they introduce
novel risks. In light of the continuing evolution in the complexity of digital mental health medical devices, regulatory approaches will also need to adapt, the FDA added, to ensure a “reasonable assurance of their safety and effectiveness while promoting innovation to support public health.”
In the meeting materials, the agency stated that although it has authorised more than 1,200 AI-enabled medical devices that span a broad range of AI technologies, none of them are indicated for mental health uses.5 Furthermore, the FDA has authorised less than 20 digital mental health medical devices that encompass non-AI technologies. To provide sponsors with insight into the current medical device landscape and regulatory expectations for AI-enabled medical devices, the agency maintains a webpage on the topic, which includes an AI-Enabled Medical Devices List.6
Digital mental health medical devices that have been authorised by the FDA to date are typically intended for prescription use and have received authorisation under several different regulations. These devices include but are not limited to computerised behavioural therapy devices for psychiatric disorders (see 21 Code of Federal Regulations [CFR] part 882.5801), digital therapy devices for attention deficit hyperactivity disorder (ADHD) (21 CFR 882.5803), digital therapy to reduce sleep disturbances for psychiatric conditions (21 CFR 882.5705), paediatric autism spectrum disorder diagnosis aid (21 CFR 882.1491) and attention task performance recorder (unclassified).
At the November DHAC meeting, the panel was presented with a medical scenario followed by discussion questions. The scenario described a patient diagnosed with major depressive disorder (MDD) by their HCP who is experiencing intermittent tearfulness due to increasing life stressors. Although the patient has consistently declined recommendations for therapy from their HCP, they are willing to try a software device that provides therapy. The device is a prescription therapy device built on a large language model (LLM) that uses contextual understanding and language generation with unique outputs that mimic a conversation with a human therapist. Its indication for use is as a standalone prescription digital therapy device indicated to treat MDD for adult patients aged 22 years and older with MDD who are not currently engaged in therapy.
There was no voting at the DHAC meeting, but the overall sentiment among panel members was one of concern. Topics highlighted by the panel included:
• Data privacy, including clarity on who owns the data gathered by the device.
• Risks of over-reliance on a machine, particularly given that the use of mobile apps on smartphones has been cited as a contributing factor in the current mental health crisis in the US.
• The need for risk-mitigation strategies to ensure users can access human support when needed.
• Mechanisms for providing feedback to the prescribing physician so they can monitor patient progress.
• Clear processes for tracking and reporting adverse events associated with device use.
• Labelling that explicitly indicates the device is AI-based.
• Requirements for long-term safety data from randomised controlled trials and strong evidence that the device does not harm people if used by unintended populations.
The FDA seeks to leverage feedback from the DHAC to help the agency foster innovation in genAI-enabled digital mental health medical devices while ensuring the safety of patients.
REFERENCES
1. World mental health today: Latest data. WHO report. https://iris.who.int/ server/api/core/bitstreams/31714489-1345-4439-8b37-6cbdc52e15ca/ content
3. Key Substance Use and Mental Health Indicators in the United States: Results from the 2024 National Survey on Drug Use and Health. https:// www.samhsa.gov/data/sites/default/files/reports/rpt56287/2024-nsduhannual-national-report.pdf
4. FDA Discussion Questions for November 6, 2025, DHAC meeting. FDA webpage. https://www.fda.gov/media/189392/download
5. FDA Executive Summary for November 6, 2025, DHAC meeting. FDA webpage. https://www.fda.gov/media/189391/download
6. Artificial Intelligence–Enabled Medical Devices. FDA webpage. https:// www.fda.gov/medical-devices/software-medical-device-samd/artificialintelligence-enabled-medical-devices
Deborah Komlos
Deborah Komlos, MS, is a former Principal Content Writer/Editor for the Cortellis suite of life science intelligence solutions at Clarivate. In this role, her coverage centredon FDA advisory committee meetings, workshops and product approvals. Her previous positions have included writing and editing for magazines, newspapers, online venues and scientific journals, as well as publication layout and graphic design work.
How Global Contact Centre Support Enhances Clinical Trial Success
Redefining Participant Support Through Global Contact Centres
As clinical trials continue to expand across geographies and adopt decentralised and hybrid models, ensuring consistent communication, safety oversight and participant support has become a critical element of trial success. Sponsors are increasingly challenged to maintain continuity across diverse regulatory environments, multiple sites, and varying participant needs, all while preserving a positive and engaging participant experience. In this evolving landscape, global contact centres are emerging as a foundational component of modern trial design, providing a centralised, scalable and human-centred support model that connects participants, sites and sponsors throughout the study lifecycle.
A well-structured global contact centre serves as more than an operational convenience. When designed with clinical rigor and patient-centric principles, it becomes a strategic asset that improves participant engagement, strengthens safety oversight and enhances trial efficiency. By offering consistent, multilingual and aroundthe-clock support, global contact centres help sponsors maintain participant trust, adherence and retention across increasingly complex trial models.
This article explores how global contact centres enable a more reliable and patient-centric clinical trial experience through consistent and compliant communication, safety reporting and delivering continuity across global, multi-site studies.
The Growing Complexity of Global and Decentralised Trials
The globalisation of clinical research has accelerated rapidly over the past decade. Trials now routinely span multiple regions, cultures, languages and healthcare systems. At the same time, decentralised and hybrid trial models have introduced new modes of participation, including virtual visits, remote monitoring and digital data capture.1 While these innovations have expanded access and convenience, they have also introduced operational and communication challenges that traditional site-centric models were designed to manage.
Participants may interact with study teams less frequently in person, rely more heavily on digital tools and navigate complex administration methods and schedules from their homes. Sites, in turn, face increased administrative burdens related to participant communications, technology support and safety reporting. Sponsors must ensure that these activities remain compliant and consistent across geographies, often with limited visibility into day-to-day participant interactions.
In this context, fragmented communication channels and inconsistent support models can undermine participant confidence and contribute to protocol deviations or premature withdrawal.
A centralised global contact centre addresses these challenges by providing a single, integrated hub for participant communication, support and issue escalation.
Connecting Participants, Sites and Sponsors through a Single Support Model
At its core, a global contact centre functions as a connective layer within the clinical trial ecosystem. Rather than relying solely on sites to manage all participant interactions, the contact centre provides a centralised point of contact that complements site activities and extends support beyond traditional working hours.
Modern global contact centres are staffed by clinically trained professionals who understand trial protocols, regulatory requirements and participant needs. These teams are equipped to handle a wide range of interactions, including general inquiries, visit coordination, study clarification, safety event intake and escalation to appropriate stakeholders. By operating within a defined governance framework and standardised processes, the contact centre ensures that information is delivered consistently and accurately across all touchpoints.
This integrated model benefits all stakeholders. Participants gain access to reliable, timely and compassionate support. Sites experience reduced administrative burden and improved focus on clinical care and data collection. Sponsors benefit from greater visibility, standardised reporting and improved operational control across their trials.
Enhancing the Participant Journey from Recruitment through Completion
Participant experience plays an increasingly central role in trial success, directly influencing recruitment, enrolment and retention. A global contact centre has emerged as a critical driver of enhancing the participant journey by providing structured, proactive and responsive support at every stage of the study.
Before enrolment, contact centres can assist with eligibility questions, informed consent navigation and scheduling of initial visits. Clear and consistent communication at this stage helps set expectations, reduces confusion and builds early trust between participants and the study team. For decentralised trials, contact centre support is particularly valuable in helping participants understand technology requirements, remote procedures and study responsibilities.
Throughout the study, contact centres reinforce engagement through appointment reminders, visit follow-ups and educational support. Participants can receive clarification on study requirements, medication administration and timelines without waiting to visit the site or for home visits. This ongoing interaction helps participants feel supported and informed, reducing anxiety and improving adherence.
Importantly, contact centres also provide a human connection that complements digital tools. While mobile apps and remote monitoring devices offer efficiency, they cannot replace the reassurance of speaking with a knowledgeable person. Access to live support fosters confidence and reinforces the participant’s sense of partnership in the clinical trial journey.
Providing 24/7, Multilingual Support to Reduce Withdrawal
Global trials often involve participants across multiple time zones, languages and cultural contexts. Without continuous support, participants may struggle to resolve issues promptly, leading to frustration, disengagement, or withdrawal from the study. Global contact centres’ round-the-clock availability ensures that participants can report concerns, ask questions, or seek assistance when issues arise, rather than delaying communication until site hours. This is particularly critical for safety-related events, technology challenges, or administration questions that may affect adherence.
Multilingual support further enhances accessibility and inclusivity. Global contact centres are comprised of bilingual or multilingual native speakers who are fully tuned with local culture and regulations, while trained to balance consistency with global standards. Participants are more likely to engage and remain in a study when they can communicate in their preferred language and feel understood. Linguistically and culturally appropriate communication helps minimise misinterpretation, supports informed decision-making and strengthens trust. By reducing barriers to communication, global contact centres help mitigate dropout risk and promote sustained engagement, an outcome that directly impacts overall trial success.
Adverse Event Reporting and Unblinding Support
Safety oversight remains a cornerstone of clinical research, regardless of trial design. In decentralised and hybrid models, however, participants may not have immediate access to site staff, increasing the importance of additional outlets for reporting. Global contact centres meet exactly that need.
Clinically trained contact centre staff can detect and intake adverse event reports and route information to the safety department promptly. This ensures timely documentation, appropriate escalation and alignment with sponsor and regulatory requirements. Standardised processes and audit-ready systems further support compliance and data integrity.
In addition to adverse event reporting, contact centres can support unblinding procedures when required for participant safety. This is available 24/7, which the sites may not be able to support outside
of the operating hours. By abiding by the governance and sponsorapproved protocols, the contact centre helps ensure that unblinding decisions are executed consistently and compliantly, minimising risk to the study and protecting trial integrity.
Delivering Consistency and Continuity Across Global, Multi-site Studies
One of the most significant advantages of a global contact centre is its ability to deliver consistency across diverse trial environments. Standardised scripts, training programs and quality assurance processes help ensure that participants receive complete, accurate and aligned information, regardless of location or mode of participation.
Consistency extends beyond messaging to include documentation, escalation pathways and performance metrics. Sponsors can define global standards while allowing for localised adaptations where required by regulation or culture. This balance supports compliance while preserving flexibility and responsiveness to participants in any location and with different needs.
Continuity is equally important. As studies progress over months or years, participants benefit from interacting with a stable support structure rather than navigating changing site staff or different communication channels. A centralised contact centre provides this continuity, reinforcing trust and reliability throughout the participant’s pre to post trial journey.
For sponsors managing large portfolios or complex global trials, this model also enables scalability. One of the unique strengths of the global contact centre is having a shared staffing model. The shared staff supports multiple programs or clients with buffering capacity and can be flexibly shifted and adjusted globally to accommodate any volume surges, such as large enrolment or geographic expansion, without disrupting participant support.
Improving Trial Efficiency through Centralised Support
Beyond participant experience and safety, global contact centres contribute directly to operational efficiency. By offloading routine inquiries and coordination tasks from sites, contact centres allow site staff to focus on clinical activities and data collection. This redistribution of workload can improve site performance, particularly when the sites have resource constraints, overloads and precautionary measures during a viral surge.
Sponsors also benefit from centralised data and reporting. Contact centre interactions generate valuable insights into participant concerns, adherence challenges and operational bottlenecks. When these are captured and analysed appropriately, sponsors can reflect and make continuous improvements in their study designs. Reduced burdens in sites and insights are increasingly critical as the sponsors are adopting decentralised or hybrid clinical trials.
A Strategic Imperative for Modern Trial Design
As clinical trials become more global, decentralised and participantcentric, the need for consistent, reliable and human-centred support has never been greater. Global contact centres are no longer a peripheral service but a strategic component of modern trial infrastructure.
By connecting participants, sites and sponsors through a single, integrated support model, global contact centres enhance the participant journey, enable timely safety oversight and deliver consistency across complex trial landscapes. Their ability to provide 24/7, multilingual and clinically informed support helps reduce dropout risk, improve retention and safeguard trial integrity.
Ultimately, investing in a well-structured global contact centre enables sponsors to deliver a more resilient, compliant and patientcentric trial experience, one that supports scientific rigor while honoring the needs and expectations of participants worldwide.
REFERENCES
1. Cummings, S. R. Clinical Trials Without Clinical Sites, JAMA 181(5), 680684 (2021)
2. https://www.propharmagroup.com/thought-leadership/clinical-trialconcierge-services-balancing-patient-experience-and-commercialoutcomes, visited on 16 Jan 2026
3. https://www.ppd.com/blog/enhancing-clinical-trials-contact-centerservices, visited on 16 Jan 2026
4. Saberi, M. Hussain, O.D., & Chang, E. Past, present and future of contact centres: a literature review, Business Process Management Journal, 23(3), 574-597 (2017)
Rajul Jain
Rajul Jain, President of Medical Information (MI) at ProPharma, has over 20 years of global experience in MI, Pharmacovigilance, Technology and Program Management. With an MBA, engineering background, PMP, and certifications including AI in Healthcare (Harvard) and ACMA, Rajul brings a wealth of knowledge in her role, leading global contact centres and is passionate about operational excellence, innovation and delivering long-term client value in healthcare and pharma.
Email: rajul.jain@propharmagroup.com
Valerie Huh
Valerie Huh, Director of Global Innovation/ Implementation at ProPharma, has over 20 years of experience in the pharmaceutical and healthcare industries, specialising in global operations and process optimisation. She holds a Pharm.D and an MBA with a certificate in Business Analytics. In her current role, she leads innovation and technology enablement within Medical Information, supporting business expansion efforts and advancing strategic initiatives aimed at improving service quality, scalability and operational efficiency.
Email: valerie.huh@propharmagroup.com
Drug Discovery USA 2026
June
(MA)
ELRIG is a not-for-profit organisation with 25 years’ experience connecting the global life science and drug discovery industry through open-access, free-to-attend events.
Would you like to reach an audience of 75% pharma & Biotech companies? Exhibit with us, email sales@elrig.org for info
250+
15 speakers 20+ exhibitors
Optimising Clinical Trials for the Era of Self-Injectable Combination Products
Self administered injectable drug–device combination (DDC) products are entering a phase of accelerated therapeutic innovation. Demographic change, chronic disease trends and the push for patient centric care are converging to make self administration a practical imperative rather than a futuristic aspiration.
In the United States, the population aged over 50 years with at least one chronic disease is projected to increase by 99.5% from 71.5 million in 2020 to 142.7 million by 2050.1 As Baby Boomers and Generation X age and live longer with multiple conditions, predictable demand is emerging for therapeutic modalities that reduce the burden on health systems while enabling patients to manage complex regimens at home. Established self administration paradigms in diabetes are now informing approaches for other patient populations and different diseases.
This shift is reflected in market dynamics. The global drug-device combination products market was valued at approximately 138 billion USD in 2023 and is expected to approach 252 billion USD by 2030,2 corresponding to a compound annual growth rate of 9.0%. Within this trajectory, self administered injectable DDCs have become a keystone technology for upgrading drug delivery, improving adherence and ultimately optimising clinical outcomes. For sponsors, the logical extension of this trend is to incorporate self injectable DDCs early, at the clinical trial stage rather than only at commercialisation.
Clinical Trial Benefits of Self Injectable DDCs
Historically, single or multi use vials have been the default presentation for subcutaneous administration during preclinical and early phase trials because they offer flexibility in fill volume and dosing while developers focus on proof of concept, safety and dose finding. The growing emphasis on patient centricity and real world usability is now prompting a shift towards self-administered dosage forms, such as prefilled syringes, safety needle devices and autoinjectors.
Several biopharmaceutical companies are investing in device research and development to enhance functionality, expand compatibility with diverse molecules and introduce combination devices earlier in the development lifecycle. When integrated into clinical trials, these DDCs offer multiple advantages:
• Improved adherence and retention through self administration that reduces reliance on clinical visits and allows patients to manage dosing in their own environment.
• Greater dosage standardisation by means of prefilled presentations that deliver consistent doses and reduce human error inherent in manual preparation.
• Enhanced data integrity via devices capable of recording dosing events, including timestamps, to support pharmacokinetic and pharmacodynamic analyses.
• Closer alignment with real world use, since trials evaluate not only the pharmacology of the drug but also the performance, usability and risk profile of the device in realistic conditions.
For participants, well designed DDCs can reduce injection related anxiety, pain and procedural complexity, contributing to a more
positive trial experience and sustained engagement. For sponsors, early integration of combination products can de risk post approval use, facilitate evidence generation for regulators and payers, reduce cost and streamline the transition from trial to market.
Design and Human Factors: A Practical Challenges Checklist
The benefits of DDCs in trials are realised only when design, usability and safety are addressed systematically. Because these products are intended for patient self-administration rather than skilled medical use, human factors engineering is central to both clinical and commercial success.
Autoinjectors illustrate these considerations clearly. Ergonomically, they must be easy to grip, orient and activate across a broad demographic, including individuals with limited dexterity, reduced muscle strength, or visual impairment. Cognitive ergonomics are equally important; intuitive operation, simple two or three step procedures and clear visual, tactile and audible feedback help patients perform injections correctly and confidently.
Device-drug compatibility is another critical axis. Autoinjectors must accommodate formulation specific attributes such as viscosity, dose volume and required injection speed. High viscosity biologics, for example, demand sufficient mechanical force and robust drive systems to deliver the full dose reliably. Rigorous stability testing must demonstrate that device materials do not adversely interact with the formulation and that the combination maintains integrity over the intended shelf life.
Delivery speed should be optimised to balance patient comfort with pharmacological needs. Consistent with ISO 11608 5 expectations, the device must reliably deliver the intended dose, particularly for biologics where minor deviations can have clinically meaningful consequences. These ‘matchmaking,’ efforts underscore that the ‘combination,’ in DDC is not rhetorical; drug and device must be developed as an integrated therapeutic system.
Equally, trial design must reflect participant diversity. Sponsors need to anticipate variations in age, comorbidities, prior experience with self injection and psychological barriers such as needle phobia. Training materials, instructions for use and support mechanisms should be adapted accordingly, without compromising the overriding priority of safety.
Safety features are fundamental in this context. Needle shields, automatic retraction mechanisms, and reliable locking systems are crucial to prevent accidental activation and needlestick injuries in populations that are not healthcare professionals. Designing for safe, error resistant use is therefore not an optional enhancement but a core requirement for trial implementation.
Regulatory and Risk Management Considerations
Regulatory pathways for self injectable DDCs are inherently more complex than for drug only products, but this complexity can ultimately facilitate smoother commercialisation once addressed early. Most self administered injectables will be classified as combination products, requiring integrated submissions that cover
both drug and device components and demonstrate compatibility between formulation and device materials.
Human factors and usability studies are central to regulatory evaluation. Sponsors must show, through realistic, scenario based testing, that lay users can safely and effectively self administer the product under expected conditions of use. This encompasses the complete user journey, including packaging, labeling, instructions for use and the step by step administration process.
Robust risk management frameworks are needed to identify, assess and mitigate issues spanning drug stability, device performance and potential interactions between components. Conducting human factors work early in the development cycle allows design modifications and process improvements before large pivotal studies are launched. Regulators increasingly regard human factors and usability engineering not as adjunct activities but as integral components of a complete DDC dossier.
Manufacturability, Supply Chain and Scale Up
Effective clinical use of DDCs must be planned with eventual commercialisation in mind. Sponsors need a realistic pathway from low volume clinical supply to potentially very high commercial volumes and this pathway must be reflected in early technical and sourcing decisions.
Key Considerations Include:
• Reliable supply of device components and assemblies, with contingency plans to mitigate disruptions that could delay trials or post approval supply.
• Strategic selection between customised autoinjectors and off the shelf platforms, balancing differentiation, technical fit, time to market and cost.
• Definition of initial and target volumes to guide investments in tooling, automation and validation, avoiding over or under engineering of manufacturing assets.
Cost–benefit analyses should be conducted during development rather than deferred to the commercial stage, recognising that clinical success may rapidly escalate volume needs. As production scales, alignment of efficacy, functionality, patient usability and cost effectiveness becomes essential to sustain both clinical and economic value.
Necessity and Opportunity in Trial Design
For sponsors deciding whether to integrate self administered injectable DDCs into clinical development, the case increasingly rests on necessity rather than optional innovation. The projected rise in chronic disease prevalence, combined with resource constraints in healthcare systems, will demand scalable, patient centric delivery models that can only be fully validated if studied under realistic conditions during trials.
Adopting DDCs in clinical studies does entail front loaded investment in device development, documentation, human factors work, manufacturing readiness and training for both site staff and participants. Yet these investments yield durable dividends in trial quality, regulatory robustness and post approval success. When thoughtfully designed and executed, incorporating self administered injectable DDCs into clinical trials transforms outsized challenges into equally substantial rewards for patients, sponsors and healthcare systems alike.
Alexander Limprecht is a Senior Director of Business Development at PCI Pharma Services. Leading EMEA business development teams, he drives strategies to support biopharma companies in bringing innovative therapies to patients through integrated clinical supply, packaging, labeling, and distribution solutions. Throughout his career, he has held several commercial roles in logistics and clinical trial supply, building strong partnerships and supporting complex global clinical programs.
De-Risking FIH: Integrated Strategies for Rapid Proof-of-Concept
The transition from preclinical to clinical testing is a pivotal moment in drug development. It’s also one of the most challenging development milestones, beset by unknown risks, unexpected data and potential regulatory hurdles to overcome. Therefore, proper planning for first-in-human (FIH) studies is critical, since missteps in trial design, CMC, or regulatory planning can cost millions and stall drug development timelines.
Accordingly, it is important to apply practical strategies proven to move molecules efficiently through early clinical assessment, including innovative single ascending dose/multiple ascending dose (SAD/MAD) designs, diverse patient recruitment tactics and fundamentally sound CMC approaches. Doing so can help drug developers avoid common development pitfalls and confidently pursue a fast-tracked path to proof-of-concept (PoC) validation.
How Current Industry Trends Impact R&D Funding
Biotech’s funding challenges and currently downward trend in investment necessitate speedy, de-risked early-stage development. Citing data from FactSet, analysts from investment bank Jefferies stated that recent U.S. policies ‘aimed at gutting the agencies responsible for conducting and regulating drug research,’ have ‘exacerbated,’ funding challenges by diminishing investor confidence in industry. Additionally, fuelled in part by advances in AI and machine learning, more compound candidates are emerging out of drug discovery, creating more competition for already limited R&D funding. This paradigm forces sponsors to be more selective in the early stages of development, so they do not advance compounds that show limited promise of success – a task that is already challenging due to increasingly complex compounds often beset by formulation challenges, such as poor solubility resulting in suboptimal exposure. However, this approach has contributed to a decline in R&D productivity, despite constantly rising costs associated with drug development.
A 2010 publication exploring the R&D productivity/rising costs dynamic highlighted the high rate of attrition during Phase 2 of development. The researchers stated that only 34% of compounds that enter Phase 2 ever progress to Phase 3. As the focus of Phase 2 is efficacy, these findings reinforce the need to incorporate PoC assessments that examine signals of efficacy earlier in development, enabling quicker wins and faster fails. Subsequent studies have reinforced the 2010 publication’s findings.
Thus, it is imperative that drug developers’ de-risk and streamline decision-making as well as critical activities during Phase 1. To achieve these goals, developers need to secure conclusive data that supports fundraising and design FIH trial protocols that deliver those data on multiple fronts. Also, it is important to trim the time between the various activities involved in transitioning a molecule from preclinical testing into the clinic. This includes reducing the white space between the drug product development and manufacture (CDMO activities) and clinical dosing (CRO activities).
Enhance FIH Study Design to Bolster Phase 2 Data
An increasingly common way to enable more robust data generation
and time-savings throughout Phase 1 research is the application of hybrid approaches that tighten the gap between Phase 1 and Phase 2. In practice, this strategy comprises an effective FIH study design with meticulous attention to basic considerations, an understanding of options that could expand or enhance the data generated and, when possible, the inclusion of patients in FIH. Initial FIH trial planning considerations include:
• Consideration of the drug candidate properties to drive an appropriate formulation strategy for the FIH study and subsequent patient trials.
• Regulatory and geographic factors that must align with the developer’s go-to-market strategy.
• Preclinical data to guide numerous FIH trial decisions, such as starting dose and exposure cap.
• Safety monitoring concerns, including whether sentinel dosing will be used and which stopping criteria will be followed.
• Dose escalation pattern decisions, including the size of dose escalation steps and how SAD/MAD are interwoven (i.e., sequential or overlapping).
• Adaptive design approaches to save time and avoid costly protocol amendments by adding predefined options to the original protocol submission that allow for adjustments as the trial progresses.
• Molecule specifics to ensure an appropriate FIH trial design, as the fundamental differences between small molecules and biologics require distinct strategic approaches.
In addition to basic FIH study considerations, trial enhancement options should be applied wherever possible. These options include:
• Food effect evaluation, which has essentially become a standard inclusion in modern FIH trials, either incorporated into the SAD or as a stand-alone cohort conducted in parallel with the MAD.
• QT data collection to help drug developers collect those data early and potentially avoid running a thorough QT (TQT) study later in development (i.e., receiving a QT waiver).
• Pharmacokinetics (PK) in different populations to address differences in gender, ethnicity, and age early in development.
• Dosage form selection, as it may be appropriate to use a fitfor-purpose drug product for the initial FIH study but then incorporate formulation flexibility to bridge to an optimised drug product for patient cohorts.
• Pharmacodynamics (PD) and early data collection in healthy volunteers to support earlier proof of mechanism of action (MoA). In such cases, it may even be feasible to add a patient cohort to the FIH trial to collect those PD data, providing an opportunity to see early efficacy data as well.
• Molecule and therapeutic area-specific data, allowing for the capture of valuable information even when not strictly necessary to support specific clinical endpoints.
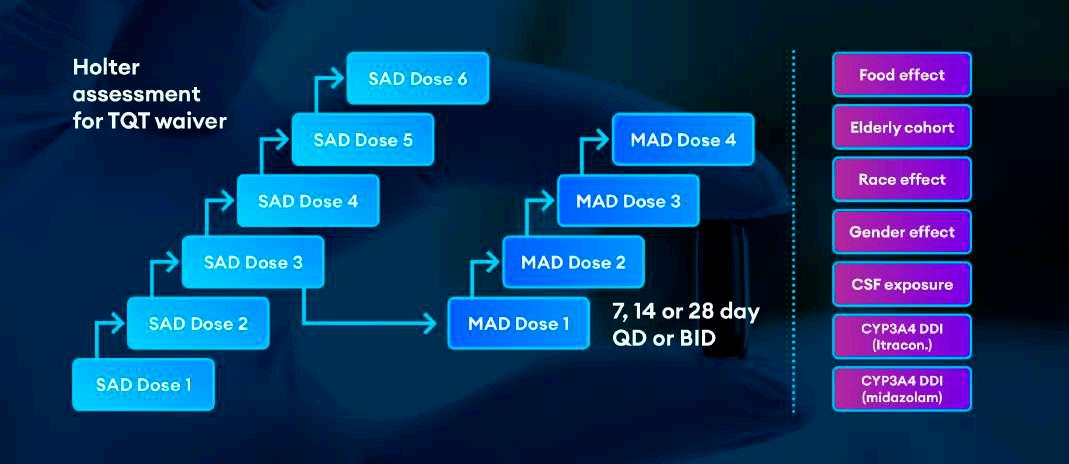
Figure 1 combines all these elements, illustrating a complete FIH trial with potential enhancements. The left side of the graphic shows a standard SAD/MAD design, as well as a Holter assessment edit for QT data collections. The right side of the graphic shows enhancements for the trial, including drug-drug interaction (DDI) assessments
for itraconazole and midazolam. Notably, this example does not include collection of any biomarkers, PD, patient data or formulation assessments.
Additionally, several different strategies can be applied or combined to include patients in early clinical studies. The most straightforward approach is direct advertising, which is most effective for prominent indications like obesity and hypertension. However, for niche therapeutic areas, drug developers may find greater success by partnering with a CRO that specialises in those specific indications. Similarly, academic collaborations can help drug developers identify early clinical trial patients for indications in, for example, rare disease. Once those patients are identified, they can be brought to a Quotient clinic location for dosing and early trial activities, or study activities can be conducted at the patient site under Quotient project management and oversight – depending on the specific circumstances and patient population.
Case Example: Accelerating Through FIH To PoC
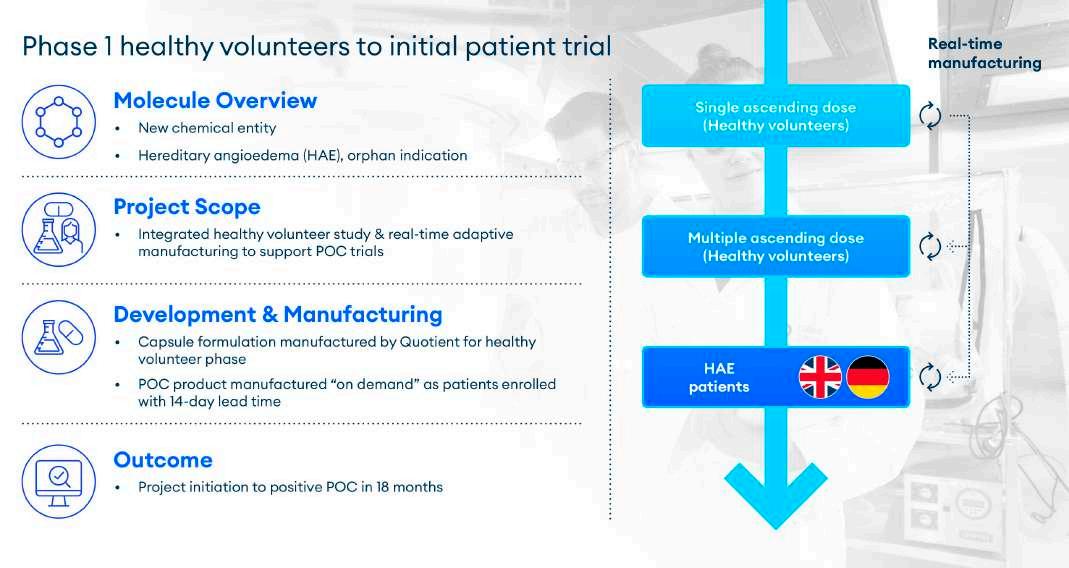
Quotient Sciences recently helped a customer navigate a new molecular entity for hereditary angioedema (HAE) through early clinical trials. As an orphan indication, patient recruitment for an HAE trial was a known challenge. To overcome this, we worked with the customer to build a program which facilitated the rapid progression of FIH assessment in healthy volunteers followed by a seamless transition into patients. The study comprised three components: SAD and MAD conducted in healthy volunteers under a single protocol and then dosing of HAE patients under a separate protocol.
The entire program was underpinned by a flexible manufacturing strategy, which enabled the FIH study to start quickly. Because of the small HAE patient numbers and difficulty in enrolment, the manufacturing supply chain needed to be capable of delivering capsule formulations for the patients in real time. In this case, a manufacturing
Figure 1
Figure 2
process was established that fulfilled a 14-day lead time for shipment to HAE patients in both the UK and in Germany.
This was accomplished using Translational Pharmaceutics®, a process by which Quotient seamlessly integrates manufacturing into the clinical supply chain, benefiting project initiation and execution by helping customers bring drugs into the clinic faster and reach decision points earlier (Figure 2).
In this case, Translational Pharmaceutics was used to support the FIH study and streamlined the process from SAD initiation to positive PoC in patients in just 18 months. This speed is possible because Quotient has simplified its integrated manufacturing and distribution processes to overcome the challenges of on-demand production. This capability is critical because drug developers need fast, reliable supply of the appropriate materials to quickly generate understanding within the Phase 1 human trial and reach FIH clinical endpoints.
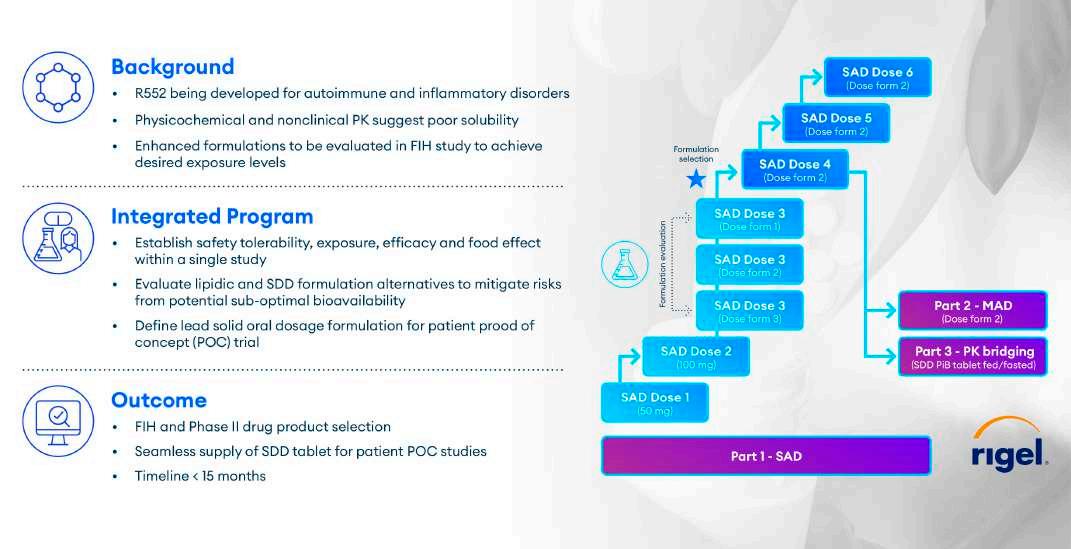
Another recent example of Translational Pharmaceutics’ effectiveness involved designing a FIH study to address suboptimal biopharmaceutic properties and preclinical data for a molecule targeting autoimmune and inflammatory disorders. Early in development, the molecule was found to have poor solubility and bioavailability due to its chemical properties. This forced the developer to manage conflicting goals: the need to advance rapidly to Phase 2 while also characterising and overcoming the compound’s perceived liabilities.
Quotient’s solution started with traditional elements of a SAD/MAD study (Figure 3). The study started dosing with an oral lipid formulation that had previously been used for toxicology studies. However, the lipid could only be given at a top dose of 180 mg in the clinic due to the RDA of the lipidic excipients. The lipid formulation was dosed in SAD 1, 2 and 3. Quotient proactively developed two additional SDD suspension formulations, with the aim to match the lipid exposure and provide linear exposure. These enhanced formulations were strategically integrated into the trial during cohort three. The two enhanced SDD formulations were assessed at the same dose level and in the same subjects as the lipid formulation in SAD cohort 3, after appropriate washout.
The blue star in Figure 3 marks the decision point after SAD dose 3 to designate an SDD formulation to progress, as well as set the dose level for SAD cohort 4. The SDD that was easier to manufacture was selected. The SAD phase continued by using the SDD to dose escalate while simultaneously being dosed in the MAD trial. After the MAD phase, researchers examined the comparative performance of the SDD as a rudimentary powder-in-bottle formulation against a final tablet formulation to bridge PK between the two.
Additionally, a fed/fasted arm was built into that element of the trial to understand potential food effects relevant to the spray dried formulation.
The study progressed quickly, advancing from FIH to the final Phase 2 drug product selection decision point in under 15 months. Further, the post-MAD comparative performance element of the trial enabled Quotient’s customer to rapidly move into Phase 2 with an optimised formulation, an SDD tablet.
Accelerating Development Through the Power of Integration
In today’s highly competitive industry, there is intense pressure on drug developers to progress studies faster into the clinical development timeline with fewer financial resources. While generating reliable data faster and more cost-effectively is inherently challenging, Quotient’s Translational Pharmaceutics® approach is proven to accelerate development programs by leveraging a purpose-built infrastructure to develop formulations, provide GMP or compounded drug products to deliver quality clinical data. Quotient’s approach has evolved to deliver integrated programs under one organisation, steered through a single project management point of contact. This is exemplified by their unique platform, Translational Pharmaceutics®, which simplifies program design and enhances decision-making on every project while also significantly reducing clients’ R&D spend. To learn more, contact the authors and visit www. quotientsciences.com/early-clinical development.
About Quotient Sciences
Quotient Sciences is a clinical development and manufacturing accelerator, helping biotech and pharma companies bring new medicines to patients faster. With over 35 years of experience and a
Figure 3
track record of success, we provide drug product (CDMO) and clinical (CRO) services across the entire development pathway, including formulation development, clinical pharmacology, clinical trial and commercial product manufacturing. Our proprietary and disruptive platform – “Translational Pharmaceutics®” – integrates drug product manufacturing and clinical testing to eliminate silos in the drug development process. This in turn reduces costs, improves outcomes and significantly accelerates drug development times.
To learn more, visit www.quotientsciences.com.
REFERENCES
1. Bell, Jacob. “Biotech Funding Plummets as Trump Policies Unnerve Investors: Jefferies.” BioPharma Dive, 4 June 2025, www.biopharmadive. com/news/biotech-funding-trump-policy-ipo-venture-pipe/749784/.
2. Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov. 2010 Mar;9(3):203-14. https://doi.org/10.1038/nrd3078. Epub 2010 Feb 19. PMID: 20168317.
3. “A Third of Clinical Trials Cancelled during Phase II.” Drug Discovery World, Drug Discovery World (DDW), 17 July 2023, www. ddw-online.com/a-thirdof-clinical-trials-cancelled-during-phase-ii-24730-202307/.
4. “Why Are Clinical Development Success Rates Falling?” Norstella, 30 Sept. 2025, www.norstella.com/why-clinical development-success-rates-falling
Dr. Andreas Reichl
Dr. Andreas Reichl has over 25 years of pharmaceutical industry experience, featuring a diverse background comprising of a medical doctorate and key positions held across clinical operations, project management and medical science. Andreas’ expertise lies in Clinical Pharmacology, where he has been advising many clients on time and cost-effective study designs, streamlined project execution and reporting. At Quotient Sciences, Andreas holds a Senior Drug Development Consultant position. Prior to his tenure at Quotient, Andreas gained extensive CRO experience at Fortrea, Labcorp Drug Development, Covance and Radiant Research, after having spent two years in academia (at University of Florida).
Dr. Kevin Schaab
Dr. Kevin Schaab has over 25 years of experience in helping develop innovative pharmaceutical therapies, with significant experience in roles spanning pharmaceutical sciences (CMC), non-clinical development, FIH to PoC clinical studies and business development. At Quotient Sciences, Kevin is a member of the drug development consultancy team and is responsible for working with our internal teams to help design and deliver early phase programs along with customer teams. Kevin holds a PhD in Chemistry from the Florida State University, a Master’s degree in Business Administration from San Diego State University, and a Bachelor of Science degree in Chemistry from the University of Cincinnati.
From Start-up to Sustainability: The New Reality of Research Site Operations
From Episodic Challenges to Structural Conditions in Clinical Research
Clinical research sites remain indispensable as the operational backbone of the clinical trial ecosystem. They are where scientific hypotheses are translated into patient/participant interactions, data collection and regulatory compliance. Yet, as identified in a recent survey of over 600 clinical research sites, sites are operating within a system under sustained and intensifying strain. Many of the pressures documented in 2025 continue in 2026, reshaping what it means for a research site to remain viable, competitive and prepared for the future.
Site readiness is no longer an episodic or transactional state. It is not something that can be switched on at study start-up and set aside at closeout. Instead, readiness has become a continuous operational condition, one that must be actively sustained amid volatility in funding, accelerating protocol complexity, proliferating technologies and persistent workforce fragility. Sites are being asked to do more, faster and with greater precision, often without proportional relief in administrative burden or structural support.
This shift has profound implications not only for sites themselves, but for sponsors and CROs who depend on their capacity. The sustainability of sites is no longer a downstream concern. It is a foundational prerequisite for the advancement of science and the timely delivery of therapies to patients.
Escalating Trial Complexity: From Challenge to Structural Feature
Among all pressures facing research sites, clinical trial complexity stands out as the most persistent and consequential. In 2025, 35% of sites identified trial complexity as their single greatest challenge, making it the most frequently cited barrier across all site types. The Tufts Centre for Drug Development (CSDD) documented increasing levels of trial complexity over the past decade, including scientific (e.g., eligibility criteria) and operational design elements (e.g., number of planned visits). While trial complexity may be considered a byproduct of innovation, it is a defining structural characteristic of modern clinical research.
Complexity manifests across multiple, interrelated dimensions. Protocols now routinely include more endpoints and procedures, resulting in more data requirements. Amendments have become more frequent, often released before a site has activated the trial. Some amendments are expected, such as those related to trial design (e.g., adaptive designs). Per the Tufts CSDD Report, on average, studies across all phases have 2.1 to 2.3 substantial amendments, with Phase II/III trials often having 2.7 to 3.5 amendments, with each requiring 6.9 changes. Operational requirements continue to expand, encompassing extensive training, layered compliance obligations and
increasingly intricate logistics. Technology demands have multiplied, with sites required to navigate multiple platforms and vendors within a single study. At the same time, data expectations have grown, with expanded requirements for collection, monitoring and reporting.
While all sites feel the effects of this escalation, the expression of complexity differs markedly by site size. Larger sites such as academic medical centres, integrated health systems and site networks often report higher absolute levels of complexity due to larger and more diverse trial portfolios, greater participation in oncology and advanced therapies and more layered internal governance structures. These sites manage scale, but at the cost of agility.
Smaller sites, including independent research centres and physician practices, experience complexity more acutely despite managing fewer studies. With fewer specialised staff, limited infrastructure redundancy and minimal capacity to absorb non-core procedures or frequent amendments, complexity translates directly into operational strain. The result across site types is a growing capacity squeeze, in which staffing, timelines and finances are stretched simultaneously, leaving little margin for error.
Study Start-up Delays: A Persistent Bottleneck in Site Readiness
Study start-up remains one of the most persistent structural bottlenecks in the clinical research lifecycle. In 2025, 31% of sites identified start-up as a top challenge, ranking it second only to trial complexity. Despite targeted process improvements over the past several years, start-up inefficiencies endure, particularly as studies become more administratively demanding.
Delays are driven by a familiar but unresolved constellation of factors: prolonged budget development and contract negotiation, increasingly complex coverage analyses and billing compliance requirements, and misalignment or slow responsiveness from sponsors and CROs. Each of these steps introduces friction; together, they create extended periods of ‘white space,’ during which sites carry cost and uncertainty without the ability to initiate patient/participant activity.
Notably, the burden of start-up delays falls unevenly across site types. Larger sites are disproportionately impacted, with 39% citing start-up as their top challenge compared with 18% of smaller sites. Decentralised research administration, multiple internal stakeholders and layered approval pathways contribute to longer timelines.
Smaller sites often move more quickly through initiation but are far more vulnerable when delays occur. Idle time can destabilise staffing models and cash flow, undermining sustainability even when eventual activation is achieved. Start-up efficiency is no longer an operational nice-to-have; it is a core component of site readiness, requiring tighter alignment across foundational elements such as
coverage analysis, budgets and clinical trial management system builds.
Workforce Shortages and Staffing Instability: A Chronic Constraint
Staffing constraints continue to exert a profound influence on site performance and resilience. In 2025, 30% of sites identified staffing as a top challenge, ranking it third overall. While some degree of staffing stabilisation was seen since the ‘Great Resignation,’ of 20212022, this issue has both broadened and deepened, extending beyond traditional coordinator and regulatory roles to encompass the broader scientific and operational workforce.
Sites report persistent difficulty recruiting and retaining qualified personnel amid rising workloads, increasingly complex protocols, and widespread burnout. Competition for talent across healthcare systems and industry has intensified, further shrinking an already constrained labor pool.
Here again, impact varies by site size. Larger sites face challenges related to workforce specialisation, unpredictable trial pipelines, and funding volatility – particularly in relation to NIH and other government sources. Smaller sites confront a different but equally destabilsing reality: limited backup capacity, heightened sensitivity to turnover, and significant operational disruption when even a single role remains unfilled.
The cumulative effect is a system operating with diminishing flexibility. Without structural intervention, workforce constraints may directly impede innovation and trial delivery, threatening not only site sustainability but the broader research enterprise.
Recruitment and Retention:
Patient/Participant Experience as a Strategic Imperative
Recruitment and retention pressures remain a continual operational barrier, cited by 28% of sites as a top challenge in 2025. While this represents a slight improvement from surveys conducted in prior
years, enrolment challenges are directly linked to participant burden, protocol design and site capacity.
Smaller sites report recruitment and retention as a more prominent challenge, with 32% citing it as a top issue. Smaller patient databases and limited brand recognition may constrain their reach, particularly as inclusion and exclusion criteria grow more restrictive.
Larger sites often recruit from broader populations but continue to face their own enrolment headwinds. Increasingly complex eligibility criteria, higher participant burden and the demands of oncology and advanced therapy trials all contribute to slower accrual and the risk of greater attrition.
Of critical note, participant experience is no longer ancillary to operational success. It is a strategic determinant of enrolment speed, retention and data quality. Sites that lack the capacity to support participants through increasingly demanding protocols face compounding challenges, potentially impacting both timelines and outcomes.
Technology Burden and Fragmentation: When Enablement Becomes Overhead
Technology adoption continues to accelerate across clinical research, yet integration and interoperability lag. In 2025, 22% of sites identified the number of technologies and vendors required for trials as the greatest driver of complexity.
The burden is particularly pronounced for smaller sites. Twentyfive percent cited difficulties with sponsor-provided technologies, compared with 17% of larger sites. Without dedicated IT support, smaller teams must rely on already stretched staff to manage training, troubleshooting and data reconciliation across multiple platforms.
The primary principle to emphasise technology should alleviate, rather than increase, the burden on sites. When evaluating new tools, ask whether they streamline existing processes or introduce
Regulatory Affairs
unnecessary complexity for site teams. Without better alignment, standardisation and integration, the promise of digital enablement risks being undermined by the operational overhead it introduces.
Funding Volatility and Financial Uncertainty: A Structural Risk
Financial volatility has become a defining feature of the site operating environment. The 2025 report details broad effects from NIH funding cuts and paused or cancelled industry trials, with ongoing uncertainty expected in 2026.
Larger sites are more likely to be affected by NIH cuts, with 24% reporting impact from both NIH funding reductions and industry pauses, compared with 9% of smaller sites. Smaller sites are more likely to report no immediate impact but express concern about future disruptions.
In 2025, sites responded to these uncertainties by reducing discretionary spending, freezing positions or reducing the number of staff, and increasing their reliance on industry-sponsored trials – strategies that may provide short-term relief but introduce longer term vulnerabilities.
Capacity
Constraints and the Redefinition of Readiness
One of the clearest indicators of systemic strain is declining site capacity to take on new studies. In 2025, 45% of sites reported that operational challenges were limiting their ability to participate in new trials. This constraint extends into 2026, reinforcing the reframing of readiness as a continuous operational asset rather than a pre-study checklist.
Nearly half of larger sites (49%) report restricted capacity, compared with 39% of smaller sites. Larger portfolios, greater administrative burden and higher exposure to funding volatility compound over time, constraining growth even in well-resourced environments.
Advanced Therapies:
The Next Wave of Complexity
In addition to prevailing challenges, the swift development of cell, gene and other advanced therapies is introducing further complexity. As of late 2025, there are over 3,200 active or planned cell and gene therapy clinical trials worldwide, with over 1,300 gene therapy candidates alone in development for cancer. Beyond these, innovative therapies in oncology, neurology/central nervous system disorders, cardiovascular and musculoskeletal diseases continue to introduce unprecedented logistical, infrastructural and safety requirements.
For sites without prior experience in these modalities, advanced therapies represent a step change in readiness requirements. Rather than replacing existing challenges, they compound them – raising the
stakes for workforce expertise, infrastructure investment and longterm follow up obligations.
Conclusion: From Managing Challenges to Building Resilience Research site challenges are no longer episodic disruptions they are structural conditions. Clinical trial complexity, staffing shortages, funding volatility, technology burden, and rising participant expectations interact in ways that amplify risk and constrain capacity.
The defining shift entering 2026 is the move from reactive problem solving toward proactive, continuous operational optimisation. Sites that endure will not simply be those that work harder, but those supported by proportional protocol design, streamlined start-up processes, integrated technologies, stable and supported workforces and true partnership with sponsors, CROs and research service providers.
The future of clinical research depends on the sustainability of its sites. Addressing these challenges is not optional. It is foundational to scientific progress, to equity in research participation and to the delivery of therapies that improve and save lives.
REFERENCES
1. 2025 Clinical Research Site Challenges Report, WCG Clinical, 2025. www. wcgclinical.com
2. WCG 2026 Trends & Insights: The Challenges and Opportunities Shaping 2026. www.wcgclinical.com
Senior Vice President, Clinical Solutions & Strategic Partnerships at WCG. Sandy works with research sites, sponsors and patient advocacy groups, supporting WCG's mission to accelerate the development of new medical therapies by improving the conduct and quality of clinical trials. She consults research sites, strategically aligning clinical solutions to improve and create efficient processes in the areas of ethical review, biosafety, trial initiation, research financial services, staffing augmentation and support for investigator-initiated trials with DSMBs and statistical consulting. She has spearheaded initiatives in research resilience with clinical trial stakeholders. Prior to WCG, Sandy led an oncology site management organisation and is currently on the Board of SASI, a non-profit research site accreditation organisation.
Ramus Medical
is a part of Ramus Corporate Group. The company is managed under a centralised quality management and has developed an integrated QMS as well as specific standard operating procedures tailored for the clinical trials department that are fully harmonised with the GCP guidelines, and the local and European legislation.
Ramus Medical EOOD is a full-service contract research organisation (CRO) in Sofia, Bulgaria.
The company was created in 2009 as a natural development of the Medical Laboratory Ramus Ltd., the largest privately-owned medical laboratory in Bulgaria.
The company independently manages clinical research projects in Bulgaria and provides partnerships in multinational clinical projects providing a comprehensive range of clinical research services:
Core Services include:
• Medical writing
Our staff has extensive expertise in the preparation, adaptation and translation of a wide range of clinical trial documents that are fully compliant with the Good Clinical Practice (GCP) standards, the client’s specifications and the regulatory requirements.
• Study start-up
We offer full or partial study start-up assistance for different types of studies throughout Bulgaria.
• Regulatory submission
• Project management
• Monitoring
• Data Management
• Pharmacokinetic evaluation
• Biostatistics
• Regulatory advice and services
• Readability User Testing
• Registration of medicinal products on the territory of Bulgaria
• Pharmacovigilance services
• Logistic department
• Destruction of IMPs/IMDs & clinical samples – agreement with PUDOOS
• Archiving services
• DDD activities
Ramus Medical has gained its expertise during the completion of numerous clinical projects carried out over the past decade:
• Phases I to IV drug trials
• Non-interventional studies
• Pilot and Pivotal Medical Device investigations
The clinical trials we conducted facilitated the MA/CE mark granted by various European Agencies/Notified Bodies and Third Country Agencies.
Ramus Medical offers flexible clinical research services in various domains, with extensive experience in fields.
Our team comprises qualified, appropriately trained, experienced, motivated and collaborative professionals and is competent to
Corporate Profile
communicate effectively across geographical and cultural boundaries to resolve any arising issues. We adhere strictly to the agreed timelines during the clinical investigations and strive to complete the tasks on time.
Why are we the solution for your projects? Ramus has its own:
Medical and Bioanalytical Laboratory
In 2018 the Medical Centre Ramus was established, located in Sofia, Bulgaria. Up to date, it has three separate locations, one of which is developed as an independent clinical research centre in compliance with the requirements for the phase I unit.
The Medical Centre Ramus allows the conduct of clinical trials in all phases in many therapeutic areas.
The Medical Centre meets all requirements for performing highquality clinical research and is designed to maximise the delivery of high-quality research data and was GCP-inspected.
Ramus Medical retains an extensive database of investigators and sites compiled through years of mutually beneficial collaboration.
Our bioanalytical laboratory is equipped with leveraging state-ofthe-art instrumentation (LC-MS/MS), techniques, and facilities, our team of experts has experience in a broad range of small molecules. Our Analytical laboratories provide method development, transfer, validation, and analysis of preclinical and clinical biological samples. We have extensive expertise in developing sensitive methods for LCMS/MS-qualifying multiple analytes and metabolites.
• Logistical company, certified for hazardous and biological samples transportation
• Clinical site facility and own catering company for hospitalised patients
Beyond the IDMC: The Value of Safety Review Committees
Continuous monitoring of safety and efficacy data in clinical trials is a fundamental component of modern clinical research. Because monitoring is complex and involves multiple stakeholders, including investigators, sponsors, contract research organisations (CROs) and other vendors, a structured and collaborative oversight process is essential to ensure timely identification and management of safety signals. Safety and efficacy review committees provide such oversight, ensuring objectivity and maintaining the scientific integrity of the clinical trial. Several types of such committees exist, with the Independent Data Monitoring Committee (IDMC), also known as the Data and Safety Monitoring Board (DSMB), being the most commonly used. The requirements for and management of IDMCs are well described in the literature; however, one type of review committee, the Safety Review Committee (SRC), has received comparatively little attention in the medical and scientific community. In this paper, we discuss key aspects of SRC composition and management.
Continuous monitoring of safety and efficacy, together with periodic risk-benefit assessments, has become an essential component of modern drug development.1 Various entities contribute to this oversight, the most widely recognised being independent data monitoring committees (IDMCs, also known as data and safety monitoring boards [DSMBs]). These entities review accumulating safety and efficacy data at predefined intervals throughout product development to provide recommendations to the sponsor on whether a trial should continue, be modified, or be stopped.2,3
In the past, drug approvals have relied on phase 2 and 3 pivotal trials. These are trials where IDMCs are typically used; however, an increasing number of recent drug approvals, particularly in oncology, are based on early-phase dose-escalation or expansion cohort clinical trials (phase 1 or 1/2). Between 2012 and 2023, these designs supported U.S. Food and Drug Administration (FDA) approval for 46 indications across 36 targeted anticancer drugs.4 For example, avapritinib, designed to treat gastrointestinal stromal tumours in patients harbouring PDGFRA exon 18 mutations, received approval in 2020 based on the results of the phase 1 NAVIGATOR trial, which enrolled 43 participants with the mutation. Similarly, avelumab, an immunotherapy drug, was approved in 2017 for treatment of metastatic Merkel cell carcinoma based on a phase 2 single-arm trial with 88 participants previously treated with chemotherapy.5–7
Phase 1 and 2 clinical trials generally enrol small numbers of participants and have limited treatment exposure and overall duration. Under these limited conditions, the use of an IDMC may be impractical. Instead, a safety review committee (SRC) can provide an efficient and proportionate mechanism for dose-escalation decisions and ongoing safety oversight.8
SRCs are already established in early-phase clinical trials (phase 1 and early phase 2), especially in dose-escalation or first-in-human (FIH) trials.9 Their main goal is to review accumulating safety, tolerability and sometimes pharmacokinetic (PK) data between
cohorts or dosing levels. The SRC determines whether it is safe to proceed to the next dose level, continue at the current dose, or pause the clinical trial.10,11
Two fundamental questions are how to distinguish between an IDMC and an SRC and whether these committees truly serve distinct functions (Table 1). Definitions provide a useful starting point, yet they reveal an important gap since multiple definitions can exist for an IDMC. For example, the FDA defines an IDMC as ‘a group of individuals with relevant expertise that reviews accumulating data on a regular basis from one or more clinical trials and recommends to the sponsor whether to continue, modify, or stop a trial or trials. A clinical trial DMC is established by the sponsor but should be independent of the sponsor and the trial conduct’.3 By contrast, no widely accepted definition exists for an SRC. Our suggestion for formally defining an SRC is ‘a group of sponsor representatives, investigator representatives and independent members as appropriate, who are responsible for reviewing ongoing safety data, including dose-limiting toxicities (DLTs), in early-phase trials to ensure participant safety and guide decisions on dose escalation and cohort progression.’ At first glance, these definitions of IDMC and SRC appear similar; however, the key distinctions lie in independence of the members and the committee’s purpose. While the members of the IDMC are external to the clinical trial under review, an SRC includes individuals actively involved in the trial. The difference is critical: the IDMC provides impartial oversight to trial participant safety and trial integrity, while the SRC focuses on ongoing monitoring and rapid response within the context of trial operations, with the added benefit of offering objective viewpoints.
Unlike IDMCs, SRCs contain study investigators and sponsor representatives. A typical SRC may include personnel such as the sponsor’s medical monitor, a clinical pharmacologist and investigators from enrolling sites. The CRO medical monitor may also participate, as they can provide a broader view of the aggregate trial data. Enrolling sites are generally defined as those with at least one participant entered in the trial, regardless of the participant’s current status, be it screening, treatment, follow-up, or discontinuation. Investigators from enrolling sites provide valuable clinical insight and firsthand knowledge of their trial participants’ conditions and the trial conduct at their site. However, investigators’ inclusion in an SRC raises concerns regarding independence and potential bias, which may affect the interpretation of safety data or decisions. To strengthen objectivity and credibility, one or two independent members external to the trial should be included. Such members enhance the integrity of safety oversight and the transparency of SRC decisions. Another reason to include an independent expert is that investigators and sponsor representatives may have limited experience in interpreting novel toxicities or rare adverse events; therefore, consultation with an external expert should be considered.
When selecting members for an SRC, the criteria should proceed as with IDMCs. For example, identify and manage potential conflicts of interest among SRC members, including independent ones. Another limitation to consider when including investigators from enrolling sites is the potential instability of committee composition. Early in the trial, the number of enrolling sites and eligible investigators may
be small, reducing diversity of opinion and possibly the quality of recommendations. Conversely, in large multicentre studies, inclusion of too many site investigators can create logistical challenges for organising SRC meetings and issuing timely recommendations. To mitigate these challenges, investigators who serve on the SRC should be preselected regardless of their sites’ enrolment status in trials with more than 10 sites. In smaller trials, the traditional approach of including investigators from enrolling sites remains feasible and should be continued.
In summary, an optimal SRC might include the sponsor’s medical monitor (or clinical lead) and a pharmacovigilance specialist, the CRO’s medical monitor (if applicable), and, depending on the size of the trial, either a limited number of selected site investigators (for larger multi-site trials) or representatives from all enrolling sites (for smaller trials with fewer than 10 sites). Ideally, one or two independent members not directly involved in the trial and external to the sponsor should be included to enhance objective oversight. A biostatistician may be invited to advise, particularly in trials employing complex statistical methods, but should serve in a non-decision-making capacity.
Developing a Charter for any type of review committee is a key element of trial governance as the Charter formally defines the committee’s roles, responsibilities, membership and operational procedures. A Charter provides consistency, transparency and accountability in safety oversight and serves as a formal reference for regulatory authorities and ethics committees.12 However, developing an SRC Charter for early-phase clinical trials poses several challenges. Defining the committee’s scope and authority can be difficult, particularly regarding dose escalation, protocol modifications, or trial suspension, especially once investigators and sponsor representatives have overlapping duties or potential conflicts of interest. Unlike an IDMC Charter, conflict of interest management depends on the composition of the SRC and is different for investigators and independent members. The Charter should also define triggers that will result in meetings – such as trial milestones, safety events, time/ enrolment-based points, dose escalation criteria and stopping rules, workflow for preparing data review packages, meeting logistics and the decision documentation process.
SRC meetings typically convene after the completion of specific study milestones, such as the enrolment or dosing of a cohort. In trials employing a traditional 3 + 3 dose-escalation design, SRC
IDMC
Typical phase of drug development
Result
Frequency
Composition
Independence
Data to be reviewed
Decisions/recommendations
Regulatory Affairs
meetings are best scheduled on an event-driven basis. For example, an optimal time for the SRC meeting to convene would be after completion of the dose limiting toxicity (DLT) observation period, which is typically 21 to 28 days following administration of the investigational product to the last participant in that cohort for each cohort, once all safety data have been verified and summarised.13,14 However, the exact timing of the SRC meeting may vary depending on the drug’s PK properties and required washout period. By that time, all relevant safety, tolerability and PK data (if applicable) should be verified, summarised and available for review. If a DLT is observed before cohort completion, an ad hoc SRC meeting may be called to determine whether to expand the cohort or suspend escalation. Meeting logistics should allow for rapid review, ideally within 72 hours of data availability and conducted via teleconference. In addition to event-driven reviews, periodic summary meetings are recommended to ensure continuous oversight of cumulative safety and PK data.15 Although the optimal frequency is not prescribed by regulatory guidance, a best-practice recommendation is to convene periodic summary meetings at predefined intervals,
SRC
Late phase (IIb–III, large pivotal trials)
Ongoing benefit–risk assessment, with the potential to recommend trial modification or termination for efficacy or safety reasons.
Periodic (e.g., quarterly or semi-annually) and/or triggered by pre-defined number of trial-specific events
Independent experts
Completely independent of the sponsor and the trial for which the IDMC was established
Blinded and unblinded cumulative, aggregated safety and/ or efficacy data in a form of tables, listings, and figures at predefined intervals
Independent recommendations to the sponsor on whether to continue, modify, or stop the trial
Early phase (I–IIa)
Immediate safety and dose-escalation decisions
Possibly after each cohort or dose step, plus additional periodic reviews at pre-defined time intervals
Investigators, the sponsor, optional: independent member(s)
Not independent (composed only of the sponsor representative and investigators) or semi-independent if includes one or several independent members
Unblinded, real-time, participant-level safety and tolerability data (e.g., extended narratives, laboratory and pharmacokinetic data)
Operational decisions on dose or cohort modification, implemented immediately following SRC deliberation
Table 1. Key Distinctions Between IDMC and SRC, The table summarizes the key distinctions between IDMCs and SRCs, highlighting that the SRC’s unblinded, investigator-inclusive composition allows for real-time operational decisions essential to early-phase dose-escalation studies.
Regulatory Affairs
about every three to six months, depending on the indication, study complexity and rate of enrolment. These periodic meetings provide an opportunity to evaluate emerging safety trends and PK profiles across cohorts, complementing cohort-specific SRC reviews. However, in early-phase, dose-escalation trials, escalation decisions should rely primarily on cohort-specific SRC evaluations following completion of each DLT observation period.
Probably the central point of any SRC’s purpose is how the data will be presented and managed for the review. In general, an SRC reviews unblinded safety data, including adverse events (AEs), serious adverse events (SAEs), clinical laboratory results, vital signs, electrocardiograms (ECGs), PK findings and relevant imaging data when supported by pharmacodynamic (PD) findings. The presented data’s scope and level of detail are determined by the study design and trial indication. Before the study begins, the sponsor should define the input from the CRO medical monitor and specify it in the SRC Charter. Data visualisation tools, such as graphical patient profiles, time-series plots of key laboratory parameters and trend analyses of AE onset and resolution can enhance interpretation and facilitate focused discussion. Unlike traditional statistical outputs for IDMC that are primarily based on aggregated tables, figures and listings, SRC reviews require participant-level data. For each participant, detailed case narratives, participant profiles and chronological presentations of safety parameters enable the committee to assess causality, emerging safety patterns and dose tolerability on an individual basis. This granular approach assists in real-time evaluation of potential DLTs and supports data-driven decisions on whether to escalate, de-escalate, or suspend dosing in treatment. This is particularly true
for phase 1, 3+3 or adaptive dose escalation designs, if the SRC is convened once 3 patients in a cohort are enrolled and treated. In such a situation, the SRC will review and discuss each trial participant separately by using an extended patient narrative for optimal data presentation.
Data files prepared for SRC review undergo standard datacleaning procedures conducted by the data management and clinical teams. Because reviews are performed while the trial is ongoing, the data may not have the completeness or verification level of a finalised study database. The SRC coordinator distributes the unblinded review package to all committee members in accordance with the procedures outlined in the SRC Charter.
Because the SRC reviews unblinded data and its membership includes the study investigators, discussions are conducted as open sessions with full access to treatment assignments and all available safety information. Decisions made at the conclusion of SRC meetings are typically final, allowing immediate implementation of recommendations regarding dose, cohort, or protocol modifications. This process enables rapid turnaround, minimising delays in study conduct while maintaining a robust safety oversight process. In contrast to IDMCs, which operate under a partially blinded model and issue formal recommendations following closed-session review and discussion, the SRC functions as an operational safety body embedded within the sponsor–investigator framework. All SRC discussions, decisions and supporting data are documented in real time to ensure transparency, traceability and compliance with regulatory expectations for ongoing safety review.
Ultimately, the effectiveness of an SRC depends on the clarity of its Charter, the precision and timeliness of its data flow, its members’ cohesion as a committee and the efficiency of its deliberations. When these components function cohesively, an SRC provides a powerful mechanism for real-time safety oversight, bridging scientific rigor and operational agility in early-phase drug development.
Conclusion
Safety Review Committees (SRCs) represent a critical component of early-phase clinical research governance. As the landscape of drug development evolves toward accelerated, adaptive and firstin-human (FIH) designs, the need for scientifically rigorous safety oversight becomes increasingly evident. Unlike independent data monitoring committees (IDMCs), which monitor the safety of trial participants through independent oversight, SRCs enable real-time, data-driven decision-making within the operational context of earlyphase studies. Their unblinded review of patient-level data allows for timely identification of emerging safety signals and supports dose modification strategies.
Establishing an effective SRC requires careful planning of its composition, procedures and data management practices. The development of an SRC Charter, clearly defining the committee’s mandate, responsibilities and decision-making processes is essential to ensure consistency and transparency. Incorporating independent experts enhances objectivity and mitigates potential bias from sponsor or investigator representatives. Standardised data visualisation tools and validated workflows further improve interpretability and expedite review timelines.
As early-phase trials increasingly provide pivotal evidence for regulatory approvals, the role of SRCs will continue to expand in scope and importance. Future guidance from regulators and professional bodies would help harmonise SRC implementation across therapeutic areas, study designs and across the healthcare industry.
REFERENCES
1. Yao B, Zhu L,Jiang Q,Xia H. Safety Monitoring in Clinical Trials. Pharmaceutics. 5. 94-106 (2013). doi:10.3390/pharmaceutics5010094.
2. Ellenberg S, Fleming T, DeMets D. Data Monitoring Committees in Clinical Trials: A Practical Perspective. New York: John Wiley & Sons; 2019
3. U.S. Food and Drug Administration. (2024, February). Use of Data Monitoring Committees in Clinical Trials: Draft guidance for industry (Revision 1) [Draft guidance]. U.S. Department of Health and Human Services. https://www.fda. gov/media/176107/download
4. Huang Y, Zhu T, Zhong J, Yuan J. Using Dose-Escalation and -Expansion Cohort Study as Pivotal Trial for Targeted Anticancer Drug Approval. JCO Precis Oncol. Aug; 9: e2500253 (2025). doi: 10.1200/PO-25-00253.
5. FDA approves avapritinib for gastrointestinal stromal tumor with a rare mutation. https://www.fda.gov/drugs/resources-information-approveddrugs/fda-approves-avapritinib-gastrointestinal-stromal-tumor-raremutation https://www.fda.gov/news-events/press-announcements/ fda-approves-first-targeted-therapy-treat-rare-mutation-patientsgastrointestinal-stromal-tumors. Visited on 17 Oct 2025.
6. Kaufman HL, Russell J, Hamid O, Bhatia S, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 17(10):1374-1385 (2016). doi: 10.1016/S1470-2045(16)30364-3.
7. Avelumab Becomes First Approved Treatment for Patients with Merkel Cell Carcinoma https://www.cancer.gov/news-events/cancer-currentsblog/2017/avelumab-fda-merkel-cell Visited on 17 Oct 2025.
8. Van Norman G. Data Safety and Monitoring Boards Should Be Required for Both Early- and Late-Phase Clinical Trials. JACC Basic Transl Sci. 6 (11):887896 (2021). doi: 10.1016/j.jacbts.2021.09.005.
9. Yap C, Solovyeva O, de Bono J, et al. Enhancing reporting quality and impact
Regulatory Affairs
of early phase dose-finding clinical trials: CONSORT Dose-finding Extension (CONSORT-DEFINE) guidance. BMJ. 383: e076387 (2023). doi: 10.1136/bmj2023-076387.
10. Dixon D, Freedman R., Herson J, et al. “Guidelines for Data and Safety Monitoring for Clinical Trials Not Requiring Traditional DMCs.” Clinical Trials. 2006;3:314-319.
11. Huang D, Dobbins D, Ghahramani P, et al. A Phase 1 Study of the Safety, Tolerability, and Pharmacokinetics of Single Ascending Doses of a First-inHuman Engineered Cationic Peptide, PLG0206, Intravenously Administered in Healthy Subjects. Antimicrob Agents Chemother. 66(1): e0144121 (2022). doi: 10.1128/AAC.01441-21.
12. DeMets D, Zarin D, Rockhold F, et al. Bringing data monitoring committee charters into the sunlight. Clin Trials. 20(4):447-451 (2023). doi: 10.1177/17407745231169499.
13. Araujo D, Greystoke A, Bates S, et al. Oncology phase I trial design and conduct: time for a change - MDICT Guidelines 2022. Ann Oncol. 34(1):4860 (2023). doi: 10.1016/j.annonc.2022.09.158.
14. Glasmacher A, Garralda E, Gwaltney C, et al. Dose optimization in cancer drug development: Review and outcome of a multi-stakeholder workshop. Eur J Cancer. 226:115593 (2025). doi: 10.1016/j.ejca.2025.115593.
15. Council for International Organizations of Medical Sciences. (2005). Management of Safety Information from Clinical Trials: Report of CIOMS Working Group VI. CIOMS. https://cioms.ch/wp-content/uploads/2017/01/ Mgment_Safety_Info.pdf
Maxim Kosov
Maxim Kosov, MD, PhD, is Senior Medical Advisor at PSI CRO AG (USA). Trained in paediatrics, anaesthesiology and intensive care, he brings more than 30 years of combined medical and research experience, including over 20 years in the clinical trials industry. His expertise spans a broad range of therapeutic areas, and he has authored or coauthored more than 60 scientific publications.
Email: maxim.kosov@psi-cro.com
Bethany Kloss
Bethany Kloss, PhD, is a Senior Medical Writer at PSI CRO AG (USA). As a research and industry scientist, she combined the fields of molecular biology, genetics and ophthalmology to investigate paediatric diseases of the eye and advanced the efforts of an international genomics sequencing facility. Moving from the discovery and preclinical stages of drug development, she has worked as a medical writer for clinical trials for the past 10 years.
Email: bethany.kloss@psi-cro.com
Jennifer Bradley
Jennifer Bradley, a Quality Control Associate at PSI CRO AG (USA). She has a BA in English from the University of Central Florida and a Medical Editing Certificate from the American Medical Writers' Association. Before providing support to Medical Writers, she worked in children's science textbook publishing, wrote training courseware for the military and wrote entertainment magazine and blog articles.
Email: jennifer.bradley@psi-cro.com
Patient-Derived Tumour Organoids for Precision Screening: Operationalising GxPLevel Quality for Translational Reliability
There is a persistent gap between preclinical promise and clinical reality in oncology drug development. Two-dimensional cell culture systems derived from animal models cannot reproduce the architectural and microenvironmental complexity that shapes therapeutic responses in human tumours. Poor clinical outcomes despite encouraging preclinical data have prompted regulatory agencies to emphasise human-relevant test systems, with reduced reliance on animal models to improve translational accuracy.
Patient-derived organoids have emerged as an alternative that replicates the three-dimensional structure, genetic diversity and heterogeneity of human tumours. However, the translational utility of these organoids depends on two inseparable pillars: biological fidelity and operational rigor. Biological fidelity ensures that in vitro responses reflect patient outcomes, while operational rigor guarantees that results are reproducible and scalable across studies and client programmes. To implement organoids from exploratory tools to standardised, decision-grade platforms requires a quality management system that is GxP-aligned and optimised for precision screening, data integration and development.
Integrating Organoids into Development and Manufacturing Workflows
In the traditional workflow, organoid-based evaluation of drug efficacy ends when a contract development and manufacturing organisation (CDMO) is selected to produce first-in-human (FIH) material. In the proposed model, organoid screening and CDMO capabilities co-exist, ensuring a seamless transition from data-driven candidate selection to commercial manufacturing.
Housing both functions within one organisation has several advantages. Results from organoid testing directly inform CDMO involvement, creating a cohesive, evidence-based handoff that strengthens early development. The integration ensures that the screening stage considers manufacturability and that the assay design considers process feasibility, formulation constraints and expression, thereby preventing technically impractical candidates from advancing.
By governing discovery and manufacturing processes, the single GxP-aligned quality system ensures reliable documentation, deviation management and data compliance, which enhances traceability and aligns with regulatory expectations. Assay parameters, growth kinetics and analytical metrics are version-controlled to streamline knowledge transfer and preserve the scientific context related to critical quality attributes and processes.
The resulting smooth transition from post-evaluation to development increases the speed to market and eliminates ambiguity.
From tissue acquisition through assay readout to manufacturing, a robust chain-of-custody ensures a unified record of the origin and analytical outcome of each model. In addition, change-control and corrective-preventive action processes manage any deviations from the model that might have downstream impacts. Operators thus consistently reverify the results when they modify any key reagents, materials, or methods.
The unified governance structure ensures that human-relevant efficacy data and manufacturability criteria serve as the basis of portfolio decisions. As a result, drug developers can prioritise the programmes that are most feasible technically and likely to succeed clinically. Ultimately, using a single quality and operational framework when conducting organoid efficacy improves data continuity, reduces late-stage rework and establishes a scientifically grounded foundation for FIH readiness.
Genomic and Transcriptomic Validation to Ensure Biological
Fidelity
The principal biological advantage of organoids lies in their capacity to reproduce tumour complexity in vitro. Their three-dimensional structure preserves cell-cell and cell-matrix interactions, which determine drug penetration, target engagement and downstream signaling. These elements are lost in two-dimensional monolayer cultures. While animal models provide in vivo contexts, they often fail to replicate human pharmacology or immune biology, limiting their predictive power. Regulatory roadmaps for New Approach Methodologies (NAMs) highlight organoids as credible, humanrelevant systems when supported by validated and quality-managed workflows. Platforms need to combine biological fidelity with GxPlevel quality controls to generate the data needed and ensure highquality outputs for regulatory approval.
Analytical rigor is a prerequisite for achieving the clinical validity and regulatory acceptance of organoid-derived data in translational contexts. Multi-omic validation of organoid models verifies biological fidelity: Whole-exome sequencing (WES) confirms that organoids retain key driver mutations and co-alterations present in the source tumour, while RNA sequencing (RNA-seq) characterises transcriptional states and pathway activities influencing the drug response. To prevent the distortion of translational relevance caused by clonal selection or culture artifacts, organoids exceeding the predefined stability thresholds are retired. Each assay is validated for dynamic range, signal-to-background ratio and intra- and interplate precision. Dose-response curves are fitted against predefined acceptance criteria, including R² thresholds and Hill slope boundaries, while reference compounds, blinded duplicates and inter-site replicates quantify reproducibility.
The proposed platform integrates WES and RNA-seq data with matched clinical and pathological datasets, enabling the accurate prediction of response and resistance. Non-responding organoids
serve as the means to identify compensatory mutations or pathway activations that undermine the primary drug mechanism. Mapping these multi-omic features to phenotypic endpoints, such as viability, apoptosis, or image-based morphology, develops composite biomarker panels that move beyond single-gene predictors. A version-controlled, quality-managed repository curates all data, creating a foundation for future machine learning (ML) models to link the genotype, phenotype and clinical context to predictive outputs. A continuous bridge between discovery and clinical translation, formed through stratified libraries, tests against models representing specific patient populations by mechanism, pathway, or chemical series.
Operationally, organoid-based screening increases portfolio efficiency by reducing false positives, which are typical in oversimplified assays. Because screening occurs in a human-tumourrelevant architecture, hits are more likely to represent true targetdriven activity, equipping drug developers with the capability to focus on candidates with greater translational potential. This approach produces leaner, stronger pipelines because fewer compounds advance, but those that do are supported by compelling evidence.
From Large-scale Screening to ML-enabled Prediction
The scale-up of organoid screening requires automation and robust data governance. Liquid-handling procedures, plating densities and assay timing must be version-controlled and environmental parameters, such as temperature and CO2 stability, must be monitored and logged. Standardised imaging pipelines that use fixed exposure and segmentation settings are key and any software updates should trigger revalidation. These measures ensure consistency and a dataset that includes all the necessary information. All primary data files must be write-protected, timestamped and access-controlled to preserve traceability and integrity. These safeguards confirm that datasets are complete and suitable for cross-study comparison, metaanalysis and potential regulatory submission.
The FDA’s roadmap for reducing animal testing underscores the importance of standardised, quality-managed organoid datasets in advancing human-relevant models. Although the current platform focuses on oncology, the same framework can be extended to other therapeutic areas once suitable tissues and endpoints are available. The GxP-aligned data architecture ensures the scale of the dataset, metadata uniformity and endpoint harmonisation, which ML depends on. Over time, validated predictive models that combine organoid phenotypes, genomic features and clinical contexts could guide trial design, dose selection and combination therapy strategies. As regulatory acceptance grows for NAMs, rigorously validated organoid data may increasingly supplement, or in some cases replace, animal data in early-stage drug development.
Bridging Current Gaps
Despite the promise of organoids, certain gaps remain. The success rate for organoid establishment varies by tumour type and time-toassays may be a limiting factor for rapidly progressing cancers. Coculture systems that incorporate immune or stromal components are under active development but are not yet standardised for large-scale application.
In addition, methodological heterogeneity between laboratories impedes cross-study data pooling. Addressing these challenges requires further standardisation, automation and multi-site validation under harmonised quality frameworks. Large prospective and harmonised studies will be essential to confirm clinical validity, demonstrate translational utility and confirm whether organoidguided therapies improve patient outcomes when compared with standard care. These solvable challenges are contingent upon collaborative infrastructure and the adherence to shared quality standards.
REFERENCES
1. U.S. Food and Drug Administration. Roadmap to reducing animal testing in preclinical safety studies.https://www.fda.gov/media/186092/download
2. Vlachogiannis G, Hedayat S, Vatsiou A, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359(6378):920-926. https://www.science.org/doi/10.1126/science.aao2774
3. Wensink GE, Elias SG, Mullenders J, et al. Patient-derived organoids as a predictive biomarker for treatment response in cancer patients. Precision Oncology. 2021;5:30. https://www.nature.com/articles/s41698-021-00168-1.pdf
4. Electronic Code of Federal Regulations. 21 CFR Part 58 — Good laboratory practice for nonclinical laboratory studies. https://www.ecfr.gov/current/ title-21/chapter-I/subchapter-A/part-58
Seahee Kim
Seahee Kim, PhD is a biopharmaceutical industry professional with over 15 years of experience across research and business strategy. At Samsung Biologics, she leads the Organoids Technology team and played a key role in the launch of Samsung Organoids, supporting oncology research by translating advanced science into strategic, clientready solutions. Prior to joining Samsung Biologics, she worked at the Samsung Advanced Institute of Technology. She received her PhD degree in Molecular Virology from Yonsei University in South Korea and post-doctoral training in Neuroscience at Penn State University in the US.
Therapeutics
Recentring Sites: The Human Factors Slowing Study Start-up and
How to Resolve Them
Despite sustained investment in technology and structured processes, study start-up timelines continue to lengthen. In a recent industry survey1 of 102 site personnel, 55% reported that activation takes five months or longer and 39% said timelines are longer than two years ago, signifying that challenges not only persist, but some may be worsening. These delays compress downstream milestones, like first patient in and last patient recruited, increasing both operational pressure and delivery risks.
If process optimisation and technology are not solving the issue, the remaining drivers are largely human, rather than technical.
Bottlenecks with Good Intentions
The clinical research industry has rightly invested in understanding and improving participant experiences. These efforts have yielded meaningful benefits for participants. However, clinical trials continue to stall at the point where participant involvement actually begins.
Rising protocol complexity and narrower eligibility criteria add to the strain sites feel at start-up. Longer start-up cycles test site capacity and readiness by increasing pressure on feasibility and recruitment planning.
These challenges are rooted less in tools than in people, expectations and workload. At the centre is a paradox: participant centricity has advanced while site centricity has lagged.
Yet sites are the operational bridge to every participant-focused ambition. When site needs are overlooked, even well-designed participant-centric strategies struggle to deliver. Sponsors and clinical research organisations therefore face a compelling conundrum: how to centre both participants and sites to improve experiences, outcomes and efficiency.
A Paradox of Centres: Balancing Participant and Site Centricity
As participant-centric strategies proliferate, they do not always account for the pressure they place on the sites that must execute them. Our survey data highlights this disconnect. Half of respondents cited complex or demanding protocols as a contributor to longer timelines and half pointed to narrow eligibility criteria as a factor extending start-up.
Refocusing on site experience creates the operational conditions required for participant-centric trial design to function in practice. Treated this way, site-centred strategy becomes a lever for faster site activation as well as stronger recruitment and retention.
The evidence points in the same direction. Sites with strong sponsor and CRO relationships tend to recruit faster and retain better. High site satisfaction correlates with clearer communication, stronger protocol adherence and more consistent participant engagement. This approach addresses many of the human factors that contribute to poor performance and start-up delays.
Dissecting Delays: Where Start-up Bottlenecks Begin Human-originated delays reflect interpersonal, procedural and
expectation-management issues rather than technology shortcomings. New tools alone will not resolve these issues. Instead, progress requires a clearer understanding of the day-to-day experiences of site personnel. Our survey findings highlight four areas for improvement:
1. Clear Communication
Only 53% rated start-up communication as good or excellent. Many sites reported confusion over timelines, responsibilities and expectations. This uncertainty erodes momentum and can dampen early enthusiasm, making it harder for sites to plan resources.
2. Protocol Clarity and Aligned Priorities
Sites are often asked to commit before receiving a final protocol or budget. Draft documents cannot replace the full detail required to make informed operational decisions under tight timelines. Unclear or shifting expectations undermine the effort sites sink into feasibility assessments and can introduce rework that lengthens cycles and contributes to a sense of burnout.
3. Administrative Strain
Rising site attrition and preselection site decline rates signal systemic strain. Preselection decline rose from 35% in 2021 to 47% in 2023. Alongside this trend, staffing shortages, administrative burden and demanding protocols stretch capacity. When sites struggle to keep pace with administrative tasks, energy shifts away from participant-facing work.
4. Contract and Budget Bottlenecks
Sequential rather than parallel negotiations slow progress and drain site resources. Nearly a quarter of respondents said they experience contract and budget delays on every study activation. Prolonged budget alignment and contract finalisation increase the risk that sites will redirect attention to other studies that are ready to move.
Across these categories the common thread is decisively human. Communication, alignment, trust and realistic expectations determine whether start-up stays on track.
Prerequisites for Recruitment and Retention
If recruitment and retention are priorities, site satisfaction must be treated as a prerequisite rather than an add on.
Positive site experience and site feasible protocols and expectations have clear effects on start-up, recruitment and retention. They shape the confidence and readiness of site teams. They influence whether a site prioritises a study in an increasingly competitive field. They determine capacity to engage and follow up with participants.
Stronger site relationships build momentum and trust at the stage where most delays and site dropouts occur. Establishing stable communication channels keeps engagement up to prevent momentum loss. Familiarity and trust accelerate decision making and reduce the waste that comes from repeated clarification cycles. These factors are especially important when sites face multiple opportunities and must choose where to invest time and resources.
In short, site satisfaction is not a soft metric. It is a leading indicator of downstream recruitment quality and retention performance. When sites feel equipped and supported, teams are more likely to sustain participant contact, guide participants through visit schedules and maintain adherence to protocol demands – all of which support fast, efficient trials from start-up onwards.
People-focused Improvements
A human-centred approach can accelerate activation without adding complexity. The following improvements are grounded in the survey findings and subsequent analysis.
1. Engage Earlier and More Meaningfully
Sites rank early involvement in protocol planning among the most impactful improvements. Early input helps balance scientific ambition with operational feasibility. It also reduces the likelihood of amendments that introduce delays and burden.
2. Streamline and Standardise Documentation
79% of respondents said simplified and standardised startup documentation would support accelerated start-up. Clear templates and consistent requirements reduce administrative load and limit rework. They also make onboarding faster for site staff who manage multiple studies.
3. Improve Alignment at Budget and Contract Stage Clearer expectations, greater transparency and parallel negotiations reduce friction and minimise dropout. Address
budgeting and contracting in tandem where possible. Set realistic timelines. Keep response loops short to preserve momentum.
These are pragmatic and people-centred changes that target the source of the delay rather than the symptoms. None requires new technology to yield benefits. All depend on better collaboration, and a shared understanding of pressure points that sites face.
Conclusion
The start-up stage is where valuable time is either gained or lost for clinical trials. Human-centred approaches can accelerate site activation by reducing friction at the point where trials often stall and by giving sites better conditions for recruitment and retention. They also reduce complexity while building trusting and supportive relationships between sites, sponsors and CROs which serves current and future studies.
The route to faster study start-up is not more technology by default. It is a clearer focus on the human factors that shape activation. Recognising site needs, improving communication and aligning expectations create conditions where participant-centric strategies can succeed. In doing so, the industry gives itself a more reliable route to better participant outcomes.
Brian Mallon is Executive Vice President, Site and Patient Solutions at ICON plc. Based in Dublin, he brings over 15 years of experience at ICON, where he has held a range of senior leadership roles across legal, procurement, and commercialisation. Prior to his current role, Brian led ICON's Commercialisation & Outcomes division, overseeing global teams in Real World Solutions, Medical Device & Diagnostic Research, and Patient Centred Services. He is known for building high-performing teams and driving innovation across operational functions that support the delivery of clinical trials.
Navigating Complexity in Master Protocols with Key Operational Partners
Master Protocols have the capacity for great advantages and efficiencies in drug development over traditional trial protocols but can be challenging for sponsors due to their inherent complexity. Involvement of experienced operational partners can help Sponsors effectively plan and execute their Master Protocols. Focusing on two types of operational partners (Trial Optimisation, Randomisation System), this paper will identify the critical role that each of these partners contribute to a Master Protocol’s adaptive design features and will provide guidance for the due diligence of operational partner selection.
What are Master Protocols?
Master Protocols (e.g., Basket, Umbrella, and Platform trial designs) are clinical trials with complex innovative designs that can offer tangible benefits to both sponsors and patients. The FDA defines a Master Protocol as ‘a protocol designed with multiple substudies, which may have different objectives and involve coordinated efforts to evaluate one or more medical products in one or more diseases or conditions within the overall study structure’.1 In comparison to traditional stand-alone trials, Master Protocols offer several advantages such as increased flexibility and efficiency in drug development, the ability to share control arms / reduced sample sizes, shared infrastructure, increased data quality and patient centricity.2 Since Master Protocols have the ability to adapt by design, this enables continuous learning and has significant advantages for patients.3 When well-planned and well-executed, Master Protocols have the potential to identify treatments that are effective or ineffective quicker than traditional trial designs. Master Protocols are particularly useful in disease states with high unmet needs.
Types of Master Protocols include:
• Basket Trials – Assessing a single therapy in multiple indications or disease populations
• Umbrella Trials – Assessing multiple therapies in a single disease population
• Platform Trials – Assessing one or more therapies, multiple phases, multiple disease populations and multiple sponsors, using a common operational framework and adaptive trial design
While Master Protocols offer a sophisticated framework for accelerating clinical development and increasing trial efficiency, several areas of challenges exist. There are regulatory challenges with the potential for complex submissions and negotiations with different agencies. There are operational challenges such as data management, drug supply, use of multiple eClinical systems and logistics. There are statistical challenges such as management of multiplicity, interim analyses, complex randomisation with adaptive design features. There can also be governance challenges with the
alignment of multiple stakeholders. Despite these challenges, the pharmaceutical industry’s growing enthusiasm for Master Protocols is likely a result of successful trials performed under such protocols in several therapeutic areas such as COVID-19, glioblastoma, oncology and amyotrophic lateral sclerosis.4
Adaptive Design Features of Master Protocols
Since Master Protocols have the ability to adapt by design, this enables continuous learning and has significant advantages for patients.3 The FDA defines an adaptive design as ‘a clinical trial design that allows for prospectively planned modifications to one or more aspects of the design based on accumulating data from subjects in the trial’.5 Thus, an adaptive design enables the trial to adapt during its course, where these adaptations are planned and specified in the trial’s protocol. Adaptations in Master Protocols are similar to standard adaptive design trials, but with additional layers of complexity. In a standard adaptive design, the same adaptations are applied to the entire trial. For instance, if an ineffective treatment were dropped, it would be omitted from the entire trial’s randomisation schedule. While in the Master Protocol framework, there are multiple layers (e.g., different subgroups, sub-protocols, sub-studies) where different adaptations can occur independently within each layer. For example, a treatment arm may be dropped in one subgroup’s randomisation, and a new treatment arm may be added in another subgroup.6
Stand-alone adaptive trial designs are complex, but even further complexity is involved with Master Protocols due to the multiple layers. Thus, to implement adaptive design features successfully, extensive planning is required.
Here are Common Adaptations in Master Protocols:
• Introducing new treatment arms (from trial or within subgroup(s))
• Dropping ineffective treatment arms (from trial or within subgroup(s))
• Pause / restart treatment arms for interim analyses (from trial or within subgroup(s))
• Treatment arm allocation ratio adjustments (from trial or within subgroup(s))
• Shared control arm across subgroups
• Change control arm if standard of care changes (from trial or within subgroup(s))
• Accounting for adaptations independently within subgroups
• Varying eligibility for treatments / subgroups (e.g., enrichment / precision medicine / biomarker-targeted)
Operational Partners
The extensive planning of the adaptive design features of a Master Protocol can be challenging for sponsors due to the inherent complexity and expertise required. Qualified operational partners can help alleviate the challenges, but the process of choosing the right operational partner can be a challenge itself. To help sponsors,
Clinical Trial Management
the Clinical Trials Transformation Initiative (CTTI) (a group focused on innovation of clinical trials co-founded by Duke University and the FDA)7) developed a robust set of resources.8 CTTI uses the terminology ‘operational partner,’ rather than ‘vendor,’ due to the critical contributions these organisations make during the development and implementation of Master Protocols.9 CTTI states that building a robust operations partner network is critical to a successful Master Protocol.9 Two types of operational partners that are essential for the planning and execution of the adaptive design features of Master Protocols are Trial Optimisation and Randomisation System. A Trial Optimisation Partner can contribute to the planning process by performing trial design simulations and recommending the most optimal designs. A Randomisation System Partner can contribute towards the planning and execution of the randomisation for that most optimal design.
Trial Optimisation Partner
While the CTTI Master Protocol operational partner assessment tool
Feature
Adding / dropping treatment arms
Response-adaptive randomisation
Shared control arm
Ability to change control arm
(if standard of care changes)
Borrowing information across treatment arms/disease categories
Simulation Capability; may require Bayesian Statistical Approach
Simulation Capability
Simulation Capability
Statistical Analysis Plan –informed by Simulation Data
Statistical Analysis Plan –informed by Simulation Data
*Information from above table based on CTTI’s Master Protocol Value Proposition Guide.9
Table 1: Master Protocol Features Requiring Simulation Capabilities
focuses on those that provide support for trial operations (e.g., data management, randomisation, site monitoring, central labs), they also have a tool dedicated to statistical simulation in Master Protocols.10 Statistical simulation capability is a prominent resource needed for several Master Protocol Features, (see Table 1). In particular, trial optimisation is a critical planning process for Master Protocols, in which high-fidelity pre-trial simulations are used to stress-test and refine adaptive trial designs before execution. These capabilities require advanced statistical expertise, specialised technology and the ability to run millions of AI-powered simulations across the design parameter space. An experienced Trial Optimisation Partner can use this process to identify the most efficient design, reduce trial timelines, optimise for statistical power and increase the probability of success, while ensuring the design meets regulatory and scientific standards. The FDA also emphasises that simulation is essential for trials such as Master Protocols with complex innovative adaptive trial designs. Simulations play a central role in determining key trial design parameters, evaluating operating characteristics and supporting productive discussions between sponsors and regulators. By exploring multiple design scenarios in silico, sponsors can address statistical, operational and regulatory considerations early which can reduce downstream risk.5,11
Simulations in Complex Adaptive Designs are Used to:
• Determine critical trial design elements
• (e.g., sample size, number and timing of interim analyses, arm allocation algorithms)
• Set thresholds for efficacy or futility, or for adding/dropping arms
• Support real-time adaptations with data-driven decisionmaking.5,11
Advanced AI and simulations capabilities such as Heterogeneous Treatment Effect (HTE) modeling and Synthetic Control Arms (SCAs) further enhance trial optimisation. HTE modeling quantifies how treatment effects vary across individuals or subgroups within a population, enabling targeted enrolment and adaptive enrichment strategies. SCAs are designed to replace or augment placebo/control arms using high-quality real-world or historical data, accelerating recruitment and reducing patient exposure to placebo. A skilled Trial Optimisation Partner can integrate these capabilities into simulation workflows, ensuring that each design iteration is both statistically robust and operationally feasible. When prior clinical trial datasets are available, a capable Trial Optimisation Partner can perform retrospective analyses to generate precise parameter estimates for simulation inputs. This ensures that trial designs are grounded in realistic assumptions, further increasing the likelihood of success.
Because the chosen Trial Optimisation Partner will directly influence timelines, cost, regulatory credibility and trial success rates, due diligence should be comprehensive. Table 2 outlines the criteria sponsors can use to evaluate potential partners. The areas for assessment should include scientific rigor, data infrastructure, simulation capabilities, operational flexibility, regulatory alignment, vendor credentials, commercial considerations and strategic fit. By considering these criteria, Sponsors will be able to compare Trial Optimisation Partners in an objective manner. They will additionally be able to address key issues related to technical rigor, data infrastructure, simulation capabilities, operational flexibility, regulatory approaches, vendor credentials, commercial considerations and strategic fit.
Clinical Trial Management
Area Criteria Assessments
Model Transparency Are the Partner’s models interpretable and explainable?
Scientific & Technical Rigor
Data Capability & Infrastructure
Methodology Look for mechanistic insights and avoid black-box ML.
Validation & Benchmarking Does the Partner use causal inference, Bayesian adaptive designs, or HTE-frameworks?
Handling of Sparse / Noisy Data
Data Breadth and Depth
Engine Input Compatibility
Has the Partner’s platform been validated against real-world clinical trial outcomes? Are external benchmarks available?
Does the Partner use data augmentation (e.g., synthetic cohorts)? Especially relevant in early-phase or rare disease modeling.
Can the Partner support ingestion of diverse data structures and formats? Are integration APIs or pipelines robust?
Privacy & Compliance Is the Partner’s platform HIPAA/GDPR-ready and compliant?
Scenario Modeling
HTE Modeling
AI & Simulation Capabilities
Operational Integration & Flexibility
Regulatory & Strategic Alignment
Synthetic Control Arms
Design Adaptation
Workflow Compatibility
Scenario Output Usability
Can the Partner’s simulations test diverse trial design elements such as entry criteria, endpoints, and recruitment rates?
Can the Partner identify subgroups (e.g., age, sex, genetic markers) with higher expected treatment benefits, potentially decreasing variance and increasing power? Can the platform learn from these insights to inform key trial design elements, such as population enrichment, stratification strategies, or mid-trial adaptation to focus on responders? HTE modeling helps identify subpopulations with different responses to the same intervention.
Does the Partner offer credible digital twin approaches? Especially important for single-arm, rare disease, and oncology settings where there are ethical or logistical barriers for placebo.
Does the Partner’s simulations support adaptive design logic (number and timing of interim analyses and decisions), or are they only for fixed design or pre-trial planning?
Can the Partner’s simulations work flexibly with the sponsor’s protocol development, CDP, or SAP workflows?
Will the Partner’s simulation output support business decisions such as trade-offs between arms, timelines, endpoints, costs? Dashboards will not do so on their own.
Speed vs Accuracy Tradeoff Are the Partner’s iteration cycles fast enough to work efficiently, yet not too fast as to compromise fidelity?
Regulatory Acceptance Have outputs from the Partner’s simulations been used in regulatory submissions (e.g., FDA Type C meetings, EMA scientific advice)?
Experience with Specific Indications
GxP/ICH Compliance
Client Base & Case Studies
Vendor Credentials
Commercial Considerations
Strategic Fit
Leadership Team & Advisors
Does the Partner understand the regulatory nuances the Sponsor is facing? Especially important in CNS, oncology, rare diseases.
Are the Partner’s simulation tools GxP/ICH Compliance? Relevant and required for simulation tools that ‘touch’ protocol designs or impact actual study execution.
What is the Partner’s track record of collaboration with established pharma/biotech companies? Are published case studies available?
Does the Partner team include people with clinical development and regulatory backgrounds or is it overly tech-centric?
Willingness to Customise Can the platform be tailored to specific trial needs or is the offering generic?
Financial & Operational Stability
IP Ownership
Pricing Models
Time to Deployment
Bridging Silos
Iterative Learning
Is the Partner financially stable and capable of long-term support?
Does the Operational Partner own the IP it uses or, alternatively, have the appropriate rights and permissions when delivering the work to you?
Does the Partner offer flexible pricing models (e.g., fixed fee, subscription, value-based)?
How long will it take to receive the first simulation results after onboarding of a Partner?
Can the Partner help bridge clinical, operational, and statistical silos or will your team need to do so on their own?
Can the Partner support iterative learning across your development pipeline – not just a one-time engagement?
Abbreviations: AI, Artificial Intelligence; APIs, Application Programming Interfaces; CDP, Clinical Development Plan; CNS, Central Nervous System; EHR, Electronic Health Record(s); EMA, European Medicines Agency; FDA, Food and Drug Administration; GDPR, General Data Protection Regulation; GxP, Good “x” Practice; HIPAA, Health Insurance Portability and Accountability Act; HTE, Heterogeneous Treatment Effects; ICH, International Council for Harmonisation; IP, Intellectual Property; ML, Machine Learning; RWD, Real-World Data; SAP, Statistical Analysis Plan
Randomisation System Partner
Table 2. Guidelines for Trial Optimisation Partner Due Diligence
With the adaptive design features required, implementing randomisation for a Master Protocol can be challenging for sponsors. Due to the complexity involved, a robust Randomisation System (e.g., web-based randomisation system (WBRS), Interactive Response Technology (IRT), Randomisation Trial Management System (RTSM)) is necessitated.6 Further, the key to a successful execution of a complex randomisation in a Master Protocol trial is the expert design of the Randomisation
System.12 Randomisation Partners should demonstrate that they have the innovative software randomisation design solutions required for Master Protocols. They should also encompass substantial experience and expertise in Master Protocols to effectively provide guidance.
Implementing randomisation for a Master Protocol is inherently more complex than a traditional clinical trial. In a traditional clinical trial, the included treatment arms and allocation ratio are the same for the entire
Clinical Trial Management
Adaptive Design Features
Introduce New Treatment Arms
Drop Ineffective Treatment Arms
Pause / Restart for Interim Analyses
Allocation Ratio Adjustments / Response Adaptive Methods
Enrichment / Precision Medicine / Biomarker-Targeted
Change to Control Arm if Standard of Care changes
Shared Control Arm
Randomisation System Implementation Requirements
Ability to add new Treatment Arms in Randomisation Schedule(s)
Ability to stop patient assignment for dropped / paused Treatment Arms; ability to restart within Randomisation Schedule(s)
Ability to adapt allocation ratio within Randomisation Schedule(s)
Ability to account for patient-level and subgroup-level treatment eligibility in Randomisation
Ability to add new Control Arm and Drop previous Control Arm from Randomisation Schedule(s)
Ability to adjust allocation ratio of involved Active Treatment Arms within Randomisation Schedule(s)
Table 3. Adaptive Design Features and Randomisation System Implementation Requirements study (e.g., often patients are randomised to one of two treatment arms in a 1:1 ratio), where a single randomisation schedule is used for patient assignments. Whereas in an adaptive design, the randomisation schedule structure may change depending on the planned adaptations (e.g., inclusion or exclusion of treatment arms, changes in allocation ratios). Thus, in this case, multiple randomisation schedules and/or flexible randomisation would be required.6 Table 3 above outlines the common adaptive design features of Master Protocols and the Randomisation System implementation requirements for flexibility.
Successful implementation of the Randomisation System expects that adaptations are executed with minimal disruptions. According to CTTI, the Randomisation Partner’s system should facilitate seamless randomisation adaptations without interruptions to functionality.9 For instance, a trial using a single fixed randomisation schedule requires a new schedule if a treatment arm is added or ratio is modified. Although this method is functional, it can be disruptive due to the time, effort and trial downtime required. A more effective solution is to incorporate flexibility into the Randomisation System, enabling realtime adaptations without the need to generate a new schedule.
CTTI recommends that the Randomisation System Partner have expertise in Master Protocol designs and dedicated biostatistics staff for effective guidance. Each Master Protocol varies, requiring different levels of flexibility.9 Adaptations (e.g., adding or dropping specific treatment arms) may be detailed in the protocol or introduced later as new treatment arms emerge. Since these arms cannot always be specified initially, the Randomisation System should allow dynamic integration of additional treatments through flexible functionality. Furthermore, any protocol-defined adaptations affecting the randomisation schedule (e.g., ratios adjusted, treatments can be paused/re-opened/dropped) should be accounted for as well. The Randomisation Partner’s experienced cross-functional and biostatistics staff should be able to assess protocols from both operational and statistical perspectives and guide the sponsor in determining which adaptations to incorporate in the system.
While it is essential for the Randomisation System Partner’s platform to possess the necessary flexibility to manage adaptations effectively, it is equally important for them to demonstrate robust capabilities to accommodate diverse randomisation methodologies. For instance, Master Protocols may employ a standard list-based randomisation design (e.g., permuted blocks with fixed treatments and allocation ratios), which is supported by most Randomisation Providers. However, certain Master Protocols might necessitate alternative randomisation approaches (e.g., Bayesian Response Adaptive Randomisation, Covariate Adaptive Randomisation/Minimisation, probabilistic assignment algorithms, or bespoke randomisation algorithms). Implementing these advanced methodologies often involves advanced software development, which can limit the number of providers that have these capabilities.13 Therefore, CTTI recommends that the Randomisation System Partner should be able to support and propose innovative software design solutions that address the requirements of advanced statistical models for randomisation.9
Collaboration of Partners
For Master Protocols to be successful, not only does an Operational Partner need to effectively collaborate with the Sponsor, but they also need to effectively collaborate with other Operational Partners. For instance, the Trial Optimisation Partner will empower data-driven decisions for the protocol’s design and execution. Whereas, the Randomisation System Partner will have to implement that design within their system while also providing consultancy on the optimal randomisation implementation. Collaboration is essential amongst these partners during the planning phase, to ensure that the protocol design and the randomisation system are effectively aligned and to establish for any communication streams and / or integrations needed for managing adaptations across the trial’s duration. Further, additional Operational Partners are often involved in the processes and integrations (e.g., Electronic Data Capture (EDC) providers for transfer of relevant patient information, patient response data).
An example that requires intense involvement and collaboration of Operational Partners is when Bayesian Response Adaptive Randomisation (RAR) is utilised. RAR is an approach that analyses accumulating patient response data through Bayesian statistical methods to dynamically adjust treatment allocation probabilities favoring better performing regimens while maintaining trial integrity and statistical validity.5 Implementing RAR within a Master Protocol brings unique computational and operational demands that extend beyond standard randomisation designs. Bayesian methods rely on extensive computations where ‘trial simulations can be particularly resource-intensive’.5 In the planning phase, the Trial Optimisation Partner will execute extensive pre-trial simulations to calibrate decision rules and control Type I error. This ensures that the RAR algorithm is developed and validated with the optimal design.
Once the RAR algorithm is designed, the Randomisation System Partner will need to collaborate with the Trial Optimisation Partner to determine how to implement the Randomisation System to align with the RAR algorithm’s parameters and protocol (i.e., any subgroups with independent probability adjustments, stratification factors). As the Randomisation System requires the RAR algorithm’s probabilities to perform treatment assignments, the partners will also need to establish how they will be transferred. Due to treatment arm additions and multiple subgroups involved with a Master Protocol, it would be operationally unfeasible to manage probability updates manually. Thus, it is necessary for the partners to establish a data integration mechanism between both systems to seamlessly transfer probability updates as they occur. The Randomisation System’s functionality then needs to be configured to perform randomisation assignments based on the current probabilities received from the Trial Optimisation System. Since RAR uses probabilities (continuous numbers) rather than ratios with integers, an approach different than a standard blocked randomisation list is required. To perform patient assignments the functionality should employ a probabilistic assignment methodology which involves utilising the RAR probabilities along with some type of random number generator and logic.13
Clinical Trial Management
To apply the RAR algorithm throughout the trial, the Trial Optimisation System needs to receive treatment arm assignment data and patient response data. Therefore, these systems also typically need to have integrations established with the EDC System where the patient responses are collected. The treatment assignment data is often needed within the EDC System from the Randomisation System, and the patient response data is needed within the Trial Optimisation System from the EDC System. The integrations and execution of the RAR algorithm address how the treatment allocation adjustments are applied across the trial. These partners will additionally have to plan out how other adaptations will be managed (e.g., add new, pause/reopen/ permanently close treatment arms). This should all be determined collaboratively during the planning phase in order for these adaptations to be performed seamlessly across the duration of the trial.
Here is a common workflow of how these integrations are applied within RAR algorithms:
• Randomisation System receives the initial probabilities from Trial Optimisation System
• Patients are assigned within the Randomisation System
• Treatment assignment data sent to EDC from Randomisation System
• EDC System collects patient responses
• EDC System sends patient response data (along with treatment assignment data received from Randomisation System) to Trial Optimisation System
• Trial Optimisation System executes RAR algorithm using most recent patient response and treatment assignment data and derives new treatment allocation probabilities
• Trial Optimisation System sends over updated probabilities to Randomisation System
• Randomisation system receives updated probabilities and uses for treatment assignment. See Figure 1 for illustration
Overall, a Master Protocol that includes RAR can be demanding. These demands include extensive pre-trial simulations to calibrate decision rules and control Type I error, high-performance computing to process interim data in real time, automated systems to update treatment allocation probabilities ratios and apply to randomisation and operational workflows to ensure data quality and rapid turnaround. For most sponsors, building and maintaining these capabilities internally is resource prohibitive. Thus, partnering with experienced Operational Partners ensures these technical and operational requirements are met seamlessly and that RAR implementation has been externally validated against gold-standard methods.
Conclusion
Overall, Master Protocols offer significant advantages in clinical trial efficiency and patient benefit but involve inherent complexity requiring expert operational support. Selecting engaged, flexible, and experienced operational partners is critical part of the planning process for sponsors. A robust operational partner network, including Trial Optimisation and Randomisation System Partners, can ensure seamless integration and successful trial execution.9 An
experienced Trial Optimisation Partner can effectively contribute through conducting AI-powered simulations to stress-test adaptive designs, identify efficient configurations, reduce timelines, optimise statistical power, and ensure regulatory compliance. Their simulation capabilities are essential for planning and managing features such as adding/dropping arms, RAR, shared control arms, changing standard of care, borrowing information across arms and enrichment strategies. Due diligence in selecting a Trial Optimisation Partner involves assessing capabilities such as scientific rigor, data infrastructure, simulation capabilities, operational flexibility, regulatory alignment, vendor credentials, commercial considerations and strategic fit. The Randomisation System Partner also plays a vital role, ‘as it uniquely sits within a clinical study’s EDC and database infrastructure’.9 The Randomisation System Partner contributes through providing consultancy on the optimal implementation for seamlessly managing the adaptive design features within the trial’s randomisation. Criteria for Randomisation System Partner selection should focus on assessing for substantial experience in Master Protocols, have platforms capable of managing/executing adaptions with flexible randomisation functionality, innovative software randomisation design solutions, in-house staff including both biostatisticians and cross-functional subject matter experts that are qualified to offer consultancy/guidance to facilitate effective implementation and ability to integrate with other key clinical systems.
Success in Master Protocols depends on collaboration between sponsors and operational partners. Collaboration across the different partners is also key for success. The Trial Optimisation Partner generates data-driven design decisions to form the optimal design. The Randomisation System Partner implements the optimal design operationally. Thus, continuous collaboration is vital for planning, design / implementation, establishing data integrations across systems and managing adaptations throughout the trial.
Jennifer Ross
Jennifer Ross is the Director of Biostatistics at Almac Clinical Technologies. She leads a group of Biostatisticians and Data Managers that specialise in randomisation design, randomisation schedules, data monitoring, simulations and consultancy for complex randomisation designs (e.g., adaptive trials, master protocols). Jennifer has over 20 years of experience in biostatistics. She has a Master’s degree in Statistics and Research Technology, and a Master’s in Psychometrics from the University of Pennsylvania.
Email: jennifer.ross@almacgroup.com
Cheryl Fitzer-Attas
Cheryl Fitzer-Attas, PhD, MBA is a pharmaceutical executive with over 20 years of experience in the life sciences industry, including clinical research, drug development and medical affairs backgrounds. Cheryl advises organisations globally and works with PhaseV as a medical director supporting biopharma sponsors and CROs with AI/ML-driven solutions for fast, efficient and accurate clinical development decisions.
Email: cheryl@phasevtrials.com
Figure 1: Data Integration Workflow applied within RAR Algorithms
Media and Communications
IPI
Peer Reviewed, IPI looks into the best practice in outsourcing management for the Pharmaceutical and BioPharmaceutical industry.
www.international-pharma.com
JCS
Peer Reviewed, JCS provides you with the best practice guidelines for conducting global Clinical Trials. JCS is the specialist journal providing you with relevant articles which will help you to navigate emerging markets.
www.journalforclinicalstudies.com
IAHJ
Peer Reviewed, IAHJ looks into the entire outsourcing management of the Veterinary Drug, Veterinary Devices & Animal Food Development Industry.
www.international-animalhealth.com
IBI
Peer reviewed, IBI provides the biopharmaceutical industry with practical advice on managing bioprocessing and technology, upstream and downstream processing, manufacturing, regulations, formulation, scale-up/technology transfer, drug delivery, analytical testing and more.
www.international-biopharma.com
PNP
Pharma Nature Positive, is a platform for all stakeholders in this industry to influence decision making by regulators, governments, investors and other service providers to achieve Nature Net Positive Results. This journal will enable pharma the ability to choose the right services to attain this goal.
www.pharmanaturepositive.com
PHARMA POD
‘Putting science into conversation, and conversation into science.’Join some of the most esteemed and integral members of the Drug Discovery & Development world as they give insights & introspect into the latest movements, discoveries and innovations within the industry.
senglobalcoms.com
Clinical Trial Management
Excel in Data Management During Clinical Trial Handoffs
Clinical trial clients often face the complex task of a contract research organisation (CRO) transition, moving critical trial functions from one CRO to a functional service provider (FSP) organisation. There are as many ways to navigate these changes as there are providers, but some best practices help to drive excellence in handoffs of data management, ensuring trials stay on track.
Such guidance supports effective transition strategies that safeguard quality and timelines. By focusing on proactive planning, collaborative culture, and rigorous oversight, a well-managed transition can solve the challenges of vendor changeovers and deliver real benefits, like sustained data integrity during handoff and uninterrupted progress in clinical trial data management. This can result in a smoother operation that mitigates risk and sets your team up for long-term success.
Overview of Best Practices for a Seamless CRO Handoff and Management
An organisation’s needs evolve and there are many reasons a client might initiate a CRO transition. Often, it’s driven by performance gaps missed timelines, high query rates, or inadequate quality from the incumbent CRO. Leadership changes or strategic shifts can also prompt a change, as can cost pressures or the need for specialised expertise. It’s rarely a single reason; most decisions to change CROs are multi-factorial. Efficiency gains and data quality improvements are often common goals.
The key is to define clear objectives at the outset, whether it’s accelerating trial execution, reducing costs, or enhancing data integrity, so that transition strategies align with tangible business outcomes. Keep in mind that changes are seldom planned far in advance, but careful preparation once the decision is made can prevent study interruptions or unreliable study data.
A successful transition starts with structured planning and strong leadership. Begin by appointing an implementation lead, who is a single point of accountability to drive the handoff process from beginning to end. This person (or team) develops a detailed implementation plan covering timelines, responsibilities and risk mitigation steps. As well as a detailed transition plan to cover all study documentation and processes implemented to date. It’s critical to map out roles and responsibilities, as well as a robust timeline.
Engage executives and the client’s leadership to collaborate on a mutually agreed plan with clear definitions. The plan should include knowledge transfer milestones for handing over documents, data and operational processes and contingency plans for any unexpected issues. It’s also recommended to structure brief overlap periods where the outgoing and incoming teams work in parallel on key tasks to guarantee nothing is missed. This level of foresight and structure ensures the transition progresses smoothly instead of chaotically.
Protecting Data Integrity and Continuity
When shifting from one vendor to another, maintaining data integrity is nonnegotiable and safeguarding the trial data is the top priority throughout. Practically, this means conducting a thorough audit
of data systems, repositories and clinical trial data management processes before the handoff. Data management experts should perform a gap analysis between the outgoing CRO’s procedures and the FSP organisation, identifying any inconsistencies in data capture, databases, or documentation.
Verify that all case report forms, databases and metadata are up to date and accessible. Address any gaps promptly and resolve any issues collaboratively before final transfer.
This diligence is critical because clients remain accountable for data quality even when work is outsourced. To manage this risk, implement strict quality control during the transition: parallel data checks, extra medical coding reviews and validation of all transferred datasets. By planning data integrity and data continuity at each step, you can avoid any lapses that could compromise the study.
One best practice is to secure access to all necessary data systems from day one of any transition. In this way, there’s no downtime in site data entry[SK2.1], cleaning, or reporting. This philosophy of no data left behind keeps the trial on sound footing.
Collaboration and One-team Culture
A seamless CRO handoff and management process isn’t just about tasks and technology it is also about people. When two essentially opposing organisations engage in the client’s trial, success hinges on creating a one-team mentality.
Place heavy emphasis on open communication and culture alignment from the very start. All stakeholders should engage as partners with a shared goal: the trial’s success. Establish regular joint meetings and status updates to keep everyone informed.
Full transparency is the rule: timelines, pending tasks, data queries and risks are openly discussed across parties. This approach builds mutual trust and prevents us vs. them dynamics. Professionalism and respect toward the outgoing CRO are also crucial because each team has value and knowledge.
Invite the outgoing team’s input and make sure to acknowledge contributions to help foster cooperation rather than defensiveness. Strive to integrate with the client’s way of working – using preferred tools and communication styles. This will enable the transition to feel as if it is evolving to a new stage. When done right, an outsider won’t even realise a vendor change has happened. A true measure of success is when you can walk into a team meeting post-transition and not distinguish who represents the client, the old CRO, or the new provider.
Risk Mitigation and Measuring Success
Any risk mitigation in vendor transitions must be proactive. While potential pitfalls are anticipated from delays in document handover to differences in SOPs, how they are addressed is maintained within a robust transition plan. For instance, if there is a risk the outgoing CRO might be slow in transferring essential documents, negotiate access to all necessary documentation ahead of time. If foreseen differences are discovered in data standards or tools, align on those early or bring in specialised support to bridge any gaps.
Clinical Trial Management
Another common risk is strained staff. Transitions can often demand extra hours from the incoming team as they ramp up. Mitigate this shift by scaling resources so that no single team member is overburdened. This helps to maintain morale and performance during the critical handoff period.
Keep a close eye on ongoing trial deliverables to ensure that database updates, patient safety monitoring and report deadlines are continuously met. By managing these risks, the goal is for zero downtime and minimal disruption.
Once the transition is complete, how does one measure its success? It’s evaluated on several levels. Look at operational metrics: Are data cleanliness and query resolution times as good as (or better than) before? Did key milestones (like database lock or interim analysis) happen on schedule after the handoff? Is the team controlling costs as projected? For example, if one original goal was to improve data quality, the query issue rates and data error trends will be monitored closely in the months after transition – a drop in data queries or faster cycle times is a clear positive indicator.
Gather feedback from the client and all team members: Do they feel the process was well-managed? Is the new team providing the expected level of service or even exceeding it? One clear sign of success is when the client’s team expresses confidence that they are now in good hands and can point to specific improvements, such as better communications or deliverables.
Internally, it is known that a transition has truly succeeded when the functional experts and the client’s stakeholders have formed into the unified team. Client satisfaction and team cohesion are as important as hard metrics.
In our experience, when both the measurable outcomes and the team sentiment are positive, the handoff can be deemed a true success. A successful transition creates value, not just avoids problems. Those are the kinds of wins that justify the effort of a transition and drive long-term excellence in clinical trials.
Melanie Dyer
Melanie Dyer, Director, Data Management, Worldwide Flex, has over 34 years of experience in data management and relationship management. Melanie is responsible for the successful execution of data management services globally across multiple therapeutic areas. Melanie’s experience with CROs and pharma companies has provided the ability to work on fullservice opportunities with a strong focus on FSP solutions. This includes support of Phase I–IV, post-market and device studies.
Kristine Smith
Kristine Smith, Associate Director, Data Management, Worldwide Flex, has 24 years in the CRO industry, including 18 years in leadership. She has managed Phase I–IV studies across oncolog – first in human, breast, lung, ovarian, solid tumours and blood cancers – and other therapeutic areas. Experienced in database builds, process improvements in data review, and database locks, Kris now oversees data management, drives process improvements and mentors her team. She is dedicated to collaboration and team success.
Measuring What Matters: Designing a Future-Fit FSP Model for Modern Drug Development
The Functional Service Provider model has grown from a tactical staffing mechanism into a strategic outsourcing framework that underpins modern clinical development. Yet, despite its evolution, the way many organisations measure FSP performance has not kept pace. For too long, success has been defined by simple inputs: how many people are on the team, how quickly they can be onboard and what their blended rates are.
These metrics were adequate when clinical pipelines were smaller, regulatory requirements were less complex and digital tools played only a supporting role. Today, however, clinical trials are more dataintensive, decentralised and globally distributed than ever before.1,2 The stakes are higher, timelines are tighter and R&D budgets face unprecedented scrutiny.
According to the Tufts Centre for the Study of Drug Development, the average cost to develop and gain approval for a new prescription medicine now exceeds $2.5 million, while development timelines frequently exceed ten years.3,4 At the same time, the cost of drug R&D has increased,5 putting sustained pressure on sponsors to extract greater value from every dollar invested. In this environment, outsourcing models can no longer be judged merely by capacity or headcount, they must deliver measurable performance, tangible efficiency gains and demonstrable value.
The Evolution of FSP in a Changing Outsourcing Landscape
Functional outsourcing has grown steadily over the past two decades. Industry analysis estimates that more than 50% of clinical development activities are now outsourced, with functional outsourcing representing one of the fastest-growing segments.6 Historically, full-service models
dominated, but sponsors are increasingly seeking hybrid and functional arrangements that preserve strategic oversight while leveraging specialised external capabilities.
Surveys of biopharma sponsors show that approximately 35% of companies have increased FSP usage in the last two years, reflecting a clear trend toward functional delivery models.7 These arrangements provide three clear advantages:
• Functional depth in specialised disciplines such as biostatistics and programming.
• Operational continuity across complex portfolios.
• Cost transparency at a functional level.
But, with the industry entering the next phase of maturity FSP models must evolve from headcount management to performance-led delivery.
The Structural Limits of a Headcount-Driven Model
Traditional FSP models scale linearly. More work requires more people. Portfolio peaks trigger resource requests. Risk mitigation often results in increasing seniority levels unnecessarily.
Over time, this creates inefficiencies:
• Senior statisticians and programmers performing repeatable production tasks.
• Rising cost structures without proportional productivity gains.
• Underutilisation of automation.
• Delivery risk accumulating quietly in complex team structures.
Linear scaling is misaligned with modern drug development, where portfolio volatility is high and submission timelines are critical. True efficiency comes not from more bodies but from smarter allocation of resources and systems.
Redefining What We Measure
A future-fit FSP model should be guided by three core questions:
• Are we delivering more with less effort?
• Is work consistently performed at the right level of expertise?
• Is automation replacing human effort rather than merely supporting it?
These questions shift the focus from staffing to system performance.
Speed: End-to-End Cycle Time
The meaningful metric is not time to hire. It is cycle time per deliverable. Sponsors should track:
• End-to-end cycle time for SDTM, ADaM and TFL outputs
• Cycle time reduction attributable to automation
• Time to scale functional capacity up or down
Even modest improvements in cycle time can have a meaningful impact on regulatory submissions and time-to-market advantage resulting in cost efficiency gains.
Quality: Right-First-Time Delivery
Project level on-time metrics are insufficient. They only flag issues after timelines are at risk. Next-generation FSP requires near realtime Right-First-Time measurements at the task and resource level. Early detection of rework trends enables proactive intervention and risk mitigation.
Cost: Evaluate Per Deliverable
Cost should be evaluated per deliverable, not per resource. Resource mix ratios are critical. The goal is not simply geographic arbitrage, but task alignment.
A meaningful KPI is a percentage of work delivered at the intended skill ratio (junior, senior, principal). This ensures senior expertise is reserved for high-value decision-making, while production work is delivered at appropriate cost levels.
THE THREE PILLARS OF A NEXT-GENERATION FSP MODEL
Pillar One: Resource Optimisation
Resource optimisation requires deliberate task allocation. Repeatable production work should not sit with high-cost senior profiles. Instead, tasks should be assigned based on:
• Task complexity
• Required expertise
• Geographic cost structure
For example:
• SDTM and ADaM production can be delivered by junior resources in lower-cost regions, supported by automation.
• Validation can sit at senior level in mid-cost regions.
• Strategic oversight and final accountability remain with principal leads.
The result is fewer total resources, stronger ownership clarity and higher-value use of senior expertise.
Pillar Two: Automation as a Structural
Level
The second key shift is making automation a structural part of the operating model. Repeatable activities such as SDTM generation, TFL production, SAP shell development and standard QC workflows should not scale through people alone, they should scale through technology.
Automation does not eliminate human oversight or accountability. Instead, it removes friction from repeatable processes, allowing teams to focus on higher-value work that requires judgment, interpretation and decision-making. When embedded thoughtfully, automation accelerates delivery, improves consistency and enables faster timelines, ultimately supporting submission readiness and strategic objectives.
This capacity-as-a-service approach avoids permanent headcount inflation while maintaining responsiveness.
Clinical Trial Management
Mobilising Outcome-Based FSP Partnerships
Designing a next-generation FSP model is only half the journey. Mobilising it across complex sponsor environments globally requires structured execution.
FSP mobilisation should be treated as an operating model transformation, not a staffing exercise. This transformation includes five key stages:
5. Scale flexibly – surge capacity adjusts with portfolio demand without destabilising the core team.
The result is a delivery ecosystem engineered for measurable, sustainable performance, not simply capacity.
Designing for What Matters
When resource optimisation, embedded automation and engineered flexibility align to outcome-based metrics, sponsors unlock durable advantages:
• Speed – reduced cycle times.
• Quality – protected senior oversight and reduced rework.
• Flexibility – capacity responsive to portfolio needs.
• Cost – structural efficiency rather than reactive reduction.
The future of FSP is not about headcount expansion. It is about intelligent ecosystem design that not only meets today’s needs but is designed to evolve with the demands of the future.
Sarah Tucker, Chief Operating Officer at Phastar, an operations leader with 25 years of experience across pharma and CRO environments. She drives operational excellence by aligning quality, compliance and delivery while building high-performing global teams. With experience spanning both sponsor and vendor settings, Sarah champions customer-centricity and believes that understanding the voice of the customer is key to delivering measurable results and sustainable growth.
The Silent Document Revolution Unblocking Clinical Trials
For all the innovation reshaping clinical research, one part of the process has remained stubbornly analog: the documents. Protocols, Statistical Analysis Plans (SAPs), Clinical Study Reports (CSRs), manuscripts, investigator brochures, amendments, regulatory packages, thousands of pages that define the scientific, operational and safety backbone of every trial. We’ve modernised data capture, cleaned up EDC workflows and automated monitoring; yet the documents that dictate what data is collected and how a study is actually run have largely been authored the same way they were twenty years ago.
That’s now changing. Quietly, but profoundly.
Documents and Data: The True Operating System of a Trial Clinical trials run on documents and data. The documents determine the data and in turn the data flows back into the documents that capture the trial operations and outcomes. This vast and complex documentation forms the scientific and operational backbone of a trial. It specifies endpoints, outlines statistical methods, dictates visit schedules and sets the assumptions that ripple through downstream systems. When these documents are slow, inconsistent, or misaligned, the entire study slows with them.
Traditionally, dozens of authors contribute to long, version-dense documents, often writing from outdated templates or conflicting drafts. Weeks are lost reconciling changes, correcting inconsistencies and fixing issues introduced through handoffs. A definition shifts slightly in one section but not another, a table misaligns with a narrative and suddenly page 22 no longer matches page 84. Anyone who has lived through a protocol review cycle knows this pain intimately.
And when inconsistencies are not detected early, the cost compounds fast. A single unaligned endpoint definition can trigger a cascade of downstream problems: CRFs must be rebuilt, data remapped, programmes rewritten, patients re-consented and entire statistical sections re-authored late in the game. By the time the study reaches submission, regulators will scrutinise every discrepancy. One misaligned definition or contradictory method section can prompt additional queries, delay approval timelines, or undermine confidence in the study’s integrity. All because the documents (the operating system of the trial) were not in harmony from the beginning.
The Breakthrough:
Purpose-built AI that Understands Clinical Trials
Repositories and stricter templates have helped, but they never solved the core problem: humans still draft everything manually. The recent leap in natural-language models, combined with domain-specific training, has finally made it possible for AI to participate directly and safely in authoring.
Several new authoring agents embed purpose-built AI trained exclusively on clinical-trial documents and vetted reference materials and validated by medical writers and biotechnicians. They understand protocols, SAPs, CSRs and myriad more document types and the structured relationships that connect them.
The system begins with a synopsis and, within hours – not weeks, produces a coherent, section-by-section draft of a protocol, SAP and other planning documents. As studies conclude, CSRs are generated from study documents and TFLs in minutes. Writers and statisticians edit in real time, while the AI continuously checks internal consistency, terminology alignment and completeness. What once required six to eight weeks now arrives as a near-submissionready draft in a few days.
Early Results: Cleaner Documents, Fewer Contradictions, Faster Starts
Organisations employing our system are reporting ~60% reductions in authoring time for core documents. But the bigger breakthrough is coherence. Because the AI understands how endpoints relate to objectives, or how SAP methodology ties back to protocol assumptions, documents read as a single narrative rather than a stitched-together patchwork.
• Objectives, endpoints and analyses stay aligned throughout.
• Tables and shells match the narrative by design.
• Review cycles shrink because reviewers find fewer discrepancies.
Writers no longer start from a blank page. They start from structure and spend their time elevating clarity, checking scientific accuracy and refining the story the data is telling.
When drafts are complete, the platform performs a wholedocument consistency sweep and flags sections requiring sign-off. It then generates downstream assets, study-building specifications, field mappings and even working tables, so that other functions like data management and biostatistics can begin immediately instead of waiting for upstream delays.
The Bigger Picture: Connected Documents Change Everything
The real transformation comes from linking documents, not just accelerating them.
In a typical study, the protocol feeds the SAP, the SAP feeds the CSR and each document depends on consistent definitions and parameters. When AI understands these dependencies:
• A machine-readable protocol can auto-populate eCRFs and build specifications.
• Statistical shells can flow directly into the CSR draft, complete with summaries and captions.
• SAPs and CSRs remain aligned with the original protocol without manual cross-checks.
This eliminates the ‘butterfly effect,’ where a small change in one document creates downstream chaos months later. It also allows data managers, programmers and statisticians to begin work earlier, compressing not only authoring time, but the entire operational startup.
Governance: The Non-negotiable Foundation AI in a regulated environment demands discipline. Our platform is built with:
• Strict hierarchies of source material.
• Complete audit trails.
• Human-in-the-loop review.
• Document-level version lineage.
The AI supports authors; it does not replace them. Final review and approval always rests with the responsible medical writer or biostatistician. And because the model is trained only on validated clinical-trial materials not generic public internet text its role is to organise, structure and align relevant evidence.
Why this Matters
Users describe the experience not only as cost savings, but as relief. A process once defined by fragmentation, fatigue and repetitive rework now feels orderly, predictable and manageable. One senior writer told us, ‘It’s like having an assistant who never gets tired and always remembers what you wrote three weeks ago.’
And while the headlines tend to focus on AI revolutionising discovery, it’s these quieter revolutions, deep inside the operational machinery that are already moving the industry forward. Better documents lead to cleaner data, smoother operations, earlier first-patient-in and ultimately, faster access to therapies for patients.
Clinical trials will not accelerate because of one big breakthrough. They will accelerate because the bottlenecks that slow them, including the humble but essential document, finally get out of the way.
This is that moment.
Fareed Melhem
Fareed Melhem is President of Veridix AI, the technology and AI division of Emmes Group. He has worked across clinical research and technology-enabled drug development, with a focus on improving how clinical trials are designed and delivered. At Veridix, he leads the application of AI across clinical research workflows to support more efficient and consistent trial execution. Earlier in his career, Fareed held senior roles at Medidata and McKinsey & Company, where he worked with global biopharma organisations on the use of digital tools in clinical development.
Next-Generation Risk-Based Monitoring: The Role of AI in Clinical Trial Oversight
Clinical trial monitoring has traditionally relied on routine onsite visits and exhaustive source data verification (SDV) to ensure patients’ safety and data integrity. While effective at detecting individual errors, this approach is resource-intensive and often limited in its ability to identify systemic risk patterns across complex trials. In response, regulatory authorities advocated for more targeted, risk-proportionate oversight models. The European Medicines Agency’s ‘Reflection Paper on Risk-Based Quality Management in Clinical Trials’ and the U.S. FDA’s guidance on ‘Oversight of Clinical Investigations – A Risk-Based Approach,’ (2013) formally endorsed these strategies. Likewise, ICH E6(R2) and the evolving E6(R3) revisions emphasise systematic risk management throughout the trial lifecycle, integrating risk identification, assessment, control, communication and review.
To operationalise these principles, technology-enabled Risk-Based Monitoring (RBM) platforms emerged. However, early RBM tools were largely static, relying on predefined dashboards and rule-based triggers, which limited their ability to adapt to dynamic trial conditions. As clinical trials become increasingly complex with adaptive designs, decentralised data capture, wearable technologies and global operational networks, the limitations of rule-based RBM models have become evident.
Artificial Intelligence (AI), including machine learning and advanced analytics, represents a transformative evolution in monitoring. AIaugmented RBM shifts the focus from reactive detection of threshold breaches to predictive, adaptive and continuously learning risk management. By dynamically analysing large volumes of multi-source trial data, identifying subtle patterns, recalibrating risk models in real time and supporting multivariate pattern recognition, AI augments human expertise and enables proactive intervention. This evolution from manual, episodic SDV to technology-enabled RBM and now AI-augmented monitoring enhances trial efficiency, data integrity, patients’ safety and regulatory compliance, marking a paradigm shift in how modern clinical trials are conducted.
Traditional Monitoring and 100% Source Data Verification
Historically, clinical trial monitoring relied heavily on frequent onsite visits by clinical research associates (CRAs) to ensure that study data were accurate, complete and verifiable. A cornerstone of this approach was 100% Source Data Verification (SDV), in which every data point recorded in the case report form (CRF) was systematically compared against the original source documents maintained at the investigative site. These source documents typically included medical records, laboratory results, imaging reports and other clinical notes documenting patient participation and study outcomes. The primary objective of this exhaustive verification process was to safeguard data
quality, ensure data integrity, warrant regulatory compliance and protect patients’ safety throughout the trial.
While traditional monitoring with 100% SDV provided a high level of data confirmation, it was resource-intensive and operationally burdensome. Frequent site visits demanded substantial time and financial investment from sponsors and CROs, particularly in large, multicentre trials conducted across multiple regions. Moreover, this approach often emphasised transcription accuracy rather than proactive identification of systemic issues, such as protocol deviations, data trends, or emerging safety signals. As clinical trials became more complex, decentralised and global in scope, the limitations of this model became increasingly apparent, driving the adoption of more efficient and risk-proportionate monitoring strategies.
Key Limitations of Traditional Monitoring with 100% SDV:
• Delayed detection of quality issues and emerging safety signals.
• Limited ability to identify systemic risks or patterns across sites.
• High operational and financial burden.
• Significant workload for monitoring teams.
Emergence of Risk-Based Monitoring (Technology-Enabled RBM) Risk-Based Monitoring (RBM) emerged as a modern approach designed to optimise monitoring efforts by prioritising activities that have the greatest impact on patients’ safety and data quality. Regulatory authorities such as the US FDA and the EMEA encouraged the adoption of RBM frameworks, which were further supported by guidelines from the International Council for Harmonisation (ICH) through updates to Good Clinical Practice principles. RBM emphasises proactive risk assessment during trial planning, enabling sponsors and CROs to identify critical data points and processes that require focused oversight.
Under the RBM framework, monitoring activities combine centralised data review with targeted on-site visits based on predefined risk indicators. Techniques such as key risk indicators (KRIs), statistical data monitoring and centralised analytics allow sponsors and CROs to detect unusual patterns, protocol deviations, or site performance issues more efficiently. By shifting the focus from exhaustive verification to risk prioritisation, RBM enables more efficient allocation of monitoring resources while maintaining regulatory compliance and ensuring high standards of trial quality and patient safety.
However, there are some limitations of Technology-Based RiskBased Monitoring (RBM) which include:
• Dependence on Predefined Risk Indicators (KRIs): RBM systems rely on predefined Key Risk Indicators and statistical thresholds
Figure: Evolution of Clinical Trial Monitoring
established during study planning, which may not capture all potential operational or clinical risks.
• Limited Ability to Detect Complex Patterns: Rule-based analytics may fail to identify subtle, multifactorial, or emerging patterns within large and heterogeneous clinical datasets.
• Potential for Undetected Emerging Risks: Because monitoring rules are predefined, novel or unexpected risks may remain unnoticed until they become significant.
• Primarily Reactive Monitoring Approach and Limited Predictive Capability: Many RBM systems identify issues only after deviations or anomalies occur rather than predicting them in advance. Traditional RBM tools lack the advanced predictive analytics needed to anticipate operational or safety risks early in the trial.
AI-Augmented Risk-Based Monitoring
Artificial intelligence (AI) introduces a new layer of analytical capability into RBM systems. AI algorithms can process vast amounts of clinical and operational data, identify subtle patterns and detect anomalies that may signal potential risks. Advances in AI and machine learning are now driving the next evolution of clinical trial monitoring. While traditional Risk-Based Monitoring (RBM) focuses on predefined risk indicators and centralised data review, AI-augmented RBM introduces advanced analytical capabilities that can process large and complex clinical datasets in real time. By applying machine learning algorithms to diverse data sources such as electronic data capture systems (EDC), randomisation tools, laboratory results, safety databases and operational metrics, AI systems can identify patterns, correlations and anomalies that may not be easily detected through conventional monitoring methods.
One of the key advantages of AI-augmented monitoring is its ability to enable predictive risk detection rather than relying solely on retrospective analysis. Machine learning models can analyse historical trial data, site performance metrics and patient-level information to predict potential issues such as enrolment delays, protocol deviations, or data inconsistencies. These predictive insights allow sponsors and clinical operations teams to intervene earlier, reducing the likelihood of significant operational or quality issues as the trial progresses.
AI-enabled monitoring platforms can also significantly enhance centralised monitoring capabilities. By continuously analysing
incoming trial data, AI systems can dynamically generate risk scores for investigative sites, patient cohorts, or specific study parameters. These dynamic risk assessments help prioritise monitoring activities and guide targeted site visits, enablling clinical research associates and clinical project managers to focus their efforts on high-risk areas that require closer oversight. As a result, monitoring resources can be deployed more efficiently while maintaining high standards of data quality and participant safety.
Importantly, AI-augmented RBM does not replace human expertise but rather enhances the decision-making capabilities of clinical trial management team. Clinical Research Associates (CRAs), medical monitors, data reviewers and clinical project managers remain essential for interpreting AI-generated insights and making informed clinical and operational decisions. By combining advanced analytics with expert clinical judgment, AI-augmented monitoring represents a powerful approach for improving trial quality, operational efficiency and proactive risk management in modern clinical research.
AI-augmented RBM leverages machine learning, natural language processing, and advanced analytics to enhance risk detection and prediction. These core capabilities include:
Predictive Risk Modeling: Traditional Risk-Based Monitoring (RBM) platforms primarily rely on predefined thresholds and rule-based triggers. For example, a site may be flagged when serious adverse event numbers exceed a specified threshold value or patient enrolment numbers are lower than the expected number or protocol deviations surpass established limits. While these approaches help identify safety or operational or data quality issues, they typically detect problems only after deviation patterns have already occurred, making traditional RBM largely reactive. In contrast, AI-enabled RBM leverages historical and real-time trial data to identify patterns that may signal emerging risks. Machine learning models can detect early indicators of operational risks such as patient recruitment rate, CRF completion rate or data integrity issues, data inconsistencies, protocol deviations, or delays in safety reporting before they exceed predefined thresholds. By shifting from retrospective signal detection to predictive risk intelligence, AI enables earlier intervention and more proactive trial monitoring and oversight. This capability is particularly valuable in fast-enrolling Phase II and Phase III trials, where delayed detection of issues can quickly propagate across multiple investigative sites.
Multivariate Risk Pattern Detection: Traditional technology-based RBM systems typically evaluate risk indicators independently,
such as adverse event frequency, query rate, or protocol deviations, using predefined thresholds. In contrast, AI-enabled monitoring systems apply multivariate analytics to examine multiple variables simultaneously, enabling the identification of complex relationships and hidden correlations within clinical trial data. For example, patterns such as rapid patient enrolment combined with a number of protocol deviations or consistent laboratory values with unusually low variability across multiple patients, or high screen-failure rates coupled with aggressive enrolment rates may indicate potential site-level risks that rule-based systems could overlook. This multidimensional analysis enhances the sensitivity and precision of centralised monitoring, allowing earlier and more reliable identification of emerging operational or data quality issues.
Dynamic and Adaptive Risk Modeling: Traditional Risk-Based Monitoring tools typically rely on static risk models established during study initiation, where key risk indicators (KRIs) and thresholds are defined and remain largely unchanged throughout the trial. In contrast, AI-enabled monitoring systems support dynamic and adaptive risk modeling by continuously analysing evolving clinical trial data. As site performance, enrolment trends, CRF completion rate, query rate, and protocol compliance patterns change over time, the system can recalibrate risk signals and adjust monitoring priorities accordingly. This adaptive approach allows monitoring strategies to better reflect real-world trial dynamics rather than relying solely on assumptions made during protocol planning.
Scalability and Integration of Multi-Source Trial Data: AI-enabled RBM platforms are designed to process large volumes of clinical trial data from multiple sources in real time. These systems can integrate data from electronic data capture (EDC), ePRO/eCOA platforms, wearable devices, imaging systems, laboratory data and remote monitoring technologies. By automatically harmonising and analysing these diverse data streams, AI systems can manage high-velocity datasets without requiring proportional increases in manual review. This scalability makes AIbased monitoring particularly well-suited for complex and data-intensive clinical trials involving a large patient pool.
Ongoing Centralised Monitoring Instead of Periodic Review: Traditional centralised monitoring is often conducted through periodic reviews, such as weekly, bi-weekly or monthly evaluations supported by static dashboards. In contrast, AI-enabled RBM enables continuous centralised monitoring by automatically analysing new data as it becomes available in the eClinical systems. Risk scores can be updated in real time, allowing emerging anomalies to be detected earlier and appropriate alerts to be generated. This shift from periodic review to continuous monitoring and oversight improves responsiveness and reduces the time between the emergence of risks and corrective action.
Higher Cost Efficiency and Resource Optimisation: Monitoring remains one of the major cost drivers in clinical trials. While traditional RiskBased Monitoring (RBM) has reduced the frequency of on-site visits compared with legacy monitoring models, substantial manual effort is still required for centralised data review and signal interpretation. AI-enabled monitoring can automate routine analytical tasks such as anomaly detection, trend analysis and risk scoring, thereby reducing the burden on monitoring teams. This allows resources to be focused on highrisk sites or critical data elements rather than being uniformly distributed across all sites. As a result, monitoring efforts can be more efficiently targeted, potentially lowering overall study costs while maintaining or enhancing trial quality.
Improved Objectivity and Consistency in Monitoring: AI-enabled RBM helps reduce variability by applying standardised analytical models and algorithms across studies. Risk signals and scores are generated using consistent, data-driven methods, providing a more objective basis for monitoring decisions while still allowing for human oversight. This standardised approach can improve consistency in monitoring practices and strengthen the transparency and defensibility of oversight during audits and regulatory inspections.
Challenges and Limitations of AI-Augmented Risk-Based Monitoring
Despite its significant potential, the implementation of AI-Augmented
Risk-Based Monitoring (RBM) presents several practical and operational challenges. One of the primary challenges is data harmonisation and integration. AI systems must aggregate and analyse data from multiple clinical trial systems, including electronic data capture (EDC), randomisation tool (RTSM), safety databases, laboratory information systems and clinical trial management platforms etc. These systems often use different data formats, standards and data structures, which can complicate integration and require extensive data normalisation. Variability in data quality, incomplete datasets and delayed data entry can further affect the reliability and performance of AI-driven analytical models.
Another key consideration is algorithm transparency and regulatory acceptance. Many machine learning models operate as complex analytical systems, making it difficult to clearly explain how specific predictions or risk scores are generated. Regulatory authorities may require a high degree of transparency, validation and documentation to ensure that automated decision-support tools do not compromise patient safety, data integrity, or regulatory compliance. In addition, organisations may encounter technical and operational challenges when integrating AI solutions with existing legacy clinical systems that were not originally designed to support advanced analytics or continuous data processing.
Operational adoption also represents an important challenge. Successful implementation of AI-augmented RBM requires organisational readiness, including appropriate infrastructure, skilled personnel and changes in monitoring workflows. Study teams must be trained to interpret AI-generated signals and incorporate them into risk management processes without over-reliance on automated outputs. To address these challenges, sponsors and contract research organisations (CROs) should establish strong governance frameworks that include transparent algorithm validation, robust data quality management, and clearly defined human oversight. Phased implementation strategies like starting with pilot programmes and gradually expanding AI capabilities, can further facilitate the responsible and effective integration of AI technologies into clinical trial monitoring.
Conclusion
AI-augmented Risk-Based Monitoring (RBM) represents a transformative shift in clinical trial monitoring and oversight, moving beyond reactive, threshold-based approaches to predictive, adaptive and continuously learning systems. Traditional RBM relies on static risk models, periodic data review, and manual interpretation of predefined indicators, which can limit responsiveness and sensitivity to emerging risks. In contrast, AI-augmented RBM leverages historical and real-time trial data to detect subtle patterns, generate dynamic risk scores and continuously recalibrate monitoring priorities. Features such as multivariate pattern recognition, automated anomaly detection and predictive modeling enable continuous, datadriven oversight that is more precise, scalable and aligned with the operational realities of complex, modern clinical trials.
Importantly, AI does not replace clinical judgment; rather, it augments human expertise by automating routine monitoring tasks and providing actionable insights, allowing study teams to focus on high-value decision-making and strategic oversight. When implemented with robust governance, transparent algorithm validation and phased operational integration, AI-augmented RBM can enhance patients’ safety, ensure data quality, maintain data integrity, optimise resource allocation and improve regulatory inspection readiness. By combining predictive intelligence with adaptive monitoring, AI-Augmented RBM empowers sponsors and CROs to transition from reactive problem-solving to proactive risk
management, setting a new standard for efficient, high-quality and future-ready clinical trial monitoring.
REFERENCES
1. U.S. Food and Drug Administration. Guidance for Industry: Oversight of Clinical Investigations – A Risk-Based Approach to Monitoring. 2013.
2. European Medicines Agency - Reflection paper on risk based quality management in clinical trials, 2013
3. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. ICH E6(R2) and E6(R3) Good Clinical Practice Guidelines.
4. Hurley C, et al. Risk-based monitoring tools for clinical trials: A systematic review. Contemporary Clinical Trials, 2016.
5. Agrafiotis DK, et al. Risk-based monitoring of clinical trials: An integrative approach. Clinical Therapeutics, 2018.
6. Adams A, et al. Risk-Based Monitoring in Clinical Trials: 2021 Update. Therapeutic Innovation & Regulatory Science, 2023.
7. Vyas, N.R. Future of risk-based monitoring in clinical trials. International Journal of Clinical Trials, 2020.
8. VKB Parasaram. Real-Time Clinical Trial Monitoring with AI-Powered Analytics. Journal of Advances in Pharmaceutical Sciences, 2025
9. David B. Olawade et al. Artificial intelligence in clinical trials: A comprehensive review of opportunities, challenges, and future directions. International Journal of Medical Informatics, 2026
10. Rahul Aggarwal et al. The potential of artificial intelligence in clinical trials. European Journal of Clinical Investigation, 2026;
11. FDA Guidance: Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products Guidance for Industry and Other Interested Parties, January 2025
12. Ashok Ghone, The Book “Risk-Based Approach To Clinical Trial Management” Published in October 2018
13. RK Ravindran et al. Evaluating the Effectiveness of Risk-Based Monitoring and Artificial Intelligence-Driven Strategies in Clinical Trial Management: A Data-Driven Analysis. International Journal of Advanced Engineering Research and Science (IJAERS), 2026
14. Benbow JH et al. Harnessing Artificial Intelligence to Transform Clinical Trials and Cancer Care: Opportunities and Challenges. The Cancer Journal, 2025
15. Vishakha Verma et al. Clinical Trial Monitoring: An Overview of Risk-based Approach. Journal of Young Pharmacists, 2023
Ashok Ghone
Ashok Ghone, PhD, MBA, is the CEO and Founder of MedInventas, bringing nearly 25 years of experience across the pharmaceutical, medical device, and CRO industries. He brings deep expertise in global clinical research, spanning clinical operations, project management, trial execution and process innovation. Ashok has successfully led cross-functional teams across local, regional and global studies in both early- and late-phase development, as well as in multiple therapeutic areas. A recognised thought leader in clinical operations excellence, he has played a pivotal role in designing and implementing frameworks for risk-based and centralised monitoring, operational process optimisation and clinical team enablement through the use of data intelligence. At MedInventas, Ashok offers domain expertise and strategic direction for developing AI-powered eClinical systems designed to transform clinical operations, trial management, and medical writing. His vision focuses on empowering life sciences organisations and CROs with intelligent, adaptive and compliant AI ecosystems that improve quality oversight, accelerate study delivery and enhance decision-making.
Email: ashok.ghone@medinventas.com
13 MAY
Join scientists, researchers and laboratory leaders from across the pharmaceutical, biotechnology and life sciences sectors for a full day of innovation, insight and networking. Discover the technologies, equipment and services supporting drug discovery, analytical development, quality control and laboratory operations.
East Midlands Conference Centre, Nottingham
top scientific suppliers, all housed under one roof