Lesen Sie mehr unter seltenekrankheit.info


Lesen Sie mehr unter seltenekrankheit.info
„Unsere
Sophie lebt mit dem Rett-Syndrom. Ihre Eltern Sonja und Johann erzählen, was die Diagnose eines unheilbaren Gendefekts bedeutet. Ein Gespräch über Pfl ege, Herausforderungen – und die kleinen Momente, die tragen.
Seite 13
Epidermolysis bullosa
Hämophilie
Primär biliäre Cholangitis
Seltene Krebserkrankungen
Spinale Muskelatrophie
VERANTWORTLICH FÜR DEN INHALT DIESER AUSGABE:

Industry Manager Health: Kerstin Boder
Layout: Juraj Príkopa Lektorat: Joseph Lammertz
Managing Director: Bob Roemké
Fotocredits: Außer anders angegeben bei Shutterstock
Medieninhaber: Mediaplanet GmbH • Bösendorferstraße 4/23 • 1010 Wien • ATU 64759844 • FN 322799f FG Wien
Impressum: mediaplanet.com/at/impressum
Druck: Mediaprint Zeitungsdruckerei
Ges.m.b.H. & Co.KG Distribution: Der Standard Verlagsgesellschaft m.b.H
Kontakt bei Mediaplanet: +43 676 847 785 115
E-Mail: kerstin.boder@mediaplanet.com ET: 27.02.2026
Die Beiträge in der Ausgabe spiegeln die unabhängigen Meinungen der jeweiligen Interviewpartner:innen wider.
Bleiben Sie in Kontakt:
Mediaplanet Austria
@mediaplanet.austria
Ungewöhnliche Symptome, eine Kombination davon und keine ansprechende
Eine seltene Erkrankung (SE) bedeutet für betroffene Personen oft einen langen und beschwerlichen Weg bis zur richtigen Diagnose und Therapie. Von einer SE spricht man der europäischen Definition folgend, wenn weniger als eine von 2.000 Personen das spezifische Krankheitsbild aufweist. Rund fünf Prozent der Bevölkerung sind davon betroffen – in Österreich sind das rund 450.000 Menschen.
Eine Diagnose unklarer Symptome ist wichtig, um eine passende Therapie zu finden und die Situation von betroffenen
Gemeinsamer Dialog und Austausch zu seltenen Erkrankungen
Samstag, 25. April 2026 • Catamaran/ÖGB, 1020 Wien
Zielpublikum: Alle Interessierten, u.a. Betroffene seltener Erkrankungen, Angehörige, medizinisches Fachpersonal, Systempartner, Vertreter:innen aus der Pharmabranche, Medien
selten verbindet. gemeinsam bewegt.
Infos, Programm und Anmeldung:
Menschen zu verbessern. SE können sich neben den gesundheitlichen Beschwerden und Risiken auf alle Bereiche des Alltags und gesellschaftlichen Lebens auswirken und auch große Herausforderungen in der psychosozialen Versorgung darstellen. Die geringe Häufigkeit der Erkrankungen führt dazu, dass medizinisches Fachwissen, Versorgungsangebot und auch Forschung begrenzt sind. Durchschnittlich dauert es daher mehr als fünf Jahre bis zur richtigen Diagnose.
Wen kontaktiert man bei unerklärlichen, chronischen Symptomen?
Die erste Anlaufstelle bietet meist die Allgemeinmedizin oder eine Fachärztin/ein Facharzt. Als zentraler Aspekt gilt das rasche Erkennen von ungewöhnlichen Symptomen sowie richtiges Handeln. Dazu gehört auch das Wissen dieser Ärzt:innen über die spezialisierten Angebote wie zum Beispiel Expertisezentren für
Entgeltliche

seltene Erkrankungen. Im Bedarfsfall sollte durch niedergelassene (Fach-)Ärzt:innen der Kontakt zu einem spezialisierten Zentrum hergestellt werden.
Expertisezentren sind zentrale, hoch spezialisierte klinische Einrichtungen für definierte Gruppen von SE. Dort erfolgen vor allem Erstdiagnostik und Therapieeinstellung, aber auch Kontrolluntersuchungen. Die Zentren ersetzen nicht die medizinische Grundversorgung, sie sind dafür da, dass Menschen mit SE eine optimale Versorgung zukommt und bestehende Expertise sichtbar gemacht und genützt wird.
Gerade für ein vergleichsweise kleines Land wie Österreich ist die Teilhabe an internationalen Netzwerken bedeutend. Die spezialisierten Zentren für SE Österreichs sind Mitglied bei allen fachspezifischen Europäischen Referenznetzwerken (ERN). Europaweite virtuelle Befundbesprechungen unter Spezialist:innen werden so möglich und Betroffene müssen nicht mehr durch Europa reisen.
Erhöhung des Bewusstseins
Das Wissen über seltene Erkrankungen und ebenso die Wissensvermittlung nehmen eine relevante Rolle ein – sowohl in der breiten Öffentlichkeit als auch im medizinischen Fachpublikum und in der Gruppe der Jungmediziner:innen und Studierenden. Mehr Wissen
Entgeltliche Einschaltung
bedeutet höhere Gesundheitskompetenz in der gesamten Gesellschaft.
Einen wichtigen Beitrag dazu leistet der „International Rare Disease Day“, der seit 2008 jedes Jahr am letzten Tag im Februar stattfindet. Die breite Öffentlichkeit und Vertreter:innen von Politik, Industrie, Forschung und Gesundheitswesen werden mit vielen Aktionen auf das Thema aufmerksam gemacht.
Pro Rare Austria organisiert auch 2026 wieder am 25. April ein „Vernetzungstreffen“. Betroffene einer seltenen Erkrankung und alle am Thema Interessierten sind herzlich eingeladen. Gemeinsamer Dialog und Austausch sollen gefördert werden, vorgestellte Best-PracticeInitiativen dienen der Wissensweitergabe untereinander.
Pro Rare Austria – „Helpline SE“
Pro Rare Austria ist die einzige Organisation in Österreich, die als Anlaufstelle und Informationsquelle für Menschen mit seltenen Erkrankungen, deren Angehörige oder Personen auf der Suche nach einer Diagnose fungiert. Für viele Betroffene sind wir „der letzte Anker“.
Alle Anfragen werden kostenfrei bearbeitet. Unser Team bietet gerne unter anderem zu folgenden Anfragen nach Möglichkeit Vernetzung, Hilfe und Unterstützung:
• wenn der Wunsch nach
Austausch mit anderen Betroffenen besteht
• bei der oft langwierigen Suche nach einer Diagnose
• bei Fragen etwa zu Sozialleistungen, Erstattung und Systempartnern
• um Informationen zu Versorgungskontakten (Zentren, Ärzt:innen) zu erhalten
Pro Rare Austria stellt eine gemeinsame Stimme für seine aktuell mehr als 125 Mitglieder und alle Betroffenen in Österreich dar. Wir vertreten Patientenorganisationen, Selbsthilfegruppen und von einer spezifischen SE betroffene Einzelpersonen. Die Allianz bringt deren Anliegen in die politische Arbeit sowie in die Projekte und Kooperationen des Verbands ein, um Verbesserungen für Betroffene zu erzielen. Unser Ziel ist ein gleichberechtigtes Leben in der Mitte der Gesellschaft für alle Menschen mit einer seltenen Erkrankung.
NÜTZLICHES WISSEN
• Es gibt mehr als 6.000 seltene Erkrankungen.
• Eine Erkrankung gilt als selten, wenn sie weniger als eine Person von 2.000 betrifft.
• Selten und doch viele: Rund 450.000 Menschen in Österreich (rund 5 %) sind von einer seltenen Erkrankung betroffen.
Lesen Sie mehr unter: Pro Rare Austria prorare-austria.org

„Meine Hautprobleme begannen 2004“, berichtet eine Patientin. Sie litt zu dieser Zeit unter wiederkehrenden Hautausschlägen und Schmerzen. Erst Jahre später wurde bei ihr eine Mycosis fungoides diagnostiziert, eine Krebserkrankung, die in Europa weniger als einen von 110.000 Menschen betrifft.1 „Ich befand mich fast zehn Jahre lang in einer Grauzone“, erinnert sie sich. Ihre anfänglichen Symptome wurden zunächst als Ekzem erkannt. Erst ein Zufallsbefund führte zur richtigen Diagnose. Diese Geschichte ist kein Einzelfall: Der Weg bis zum Befund dauert bei diesem Krankheitsbild durchschnittlich
zwei bis sieben Jahre.2
Kyowa Kirin ist ein global tätiges biopharmazeutisches Unternehmen, das die Versorgung von Menschen mit seltenen Erkrankungen verbessern möchte. Es wurde 1949 in Japan gegründet und entwickelt seit dieser Zeit innovative Therapien in den Bereichen Nephrologie, Neurologie, Onkologie und Immunologie. Die Forschung, Entwicklung und Wirkstoffproduktion stützen sich auf Verfahren der Spitzenbiotechnologie aus eigenem Hause. So gilt das Unternehmen als Pionier in der Behandlung des nur selten auftretenden Phosphatdiabetes. Ein weiterer Schwerpunkt ist
die Behandlung seltener Krebserkrankungen wie der Mycosis fungoides und des Sézary-Syndroms – beides Unterformen des kutanen T-Zell-Lymphoms (CTCL). Kyowa Kirin möchte sämtlichen Menschen, mit denen es sich im Austausch befindet, ein Lächeln schenken – nicht nur durch die Bereitstellung neuer Wirkstoffe, sondern auch durch gelebte Partnerschaften. Das Unternehmen sucht weltweit den Austausch mit Betroffenen und Beteiligten, um gemeinsam bessere Antworten auf Patientenbedürfnisse zu finden, getrieben von dem Ansporn „Making people smile“.
Orphanet: tinyurl.com/ 4vr9ar9v 2 CL Foundation: tinyurl.com/ mvk67utw
Helpline SE Näheres unter: www.kyowakirin international.com
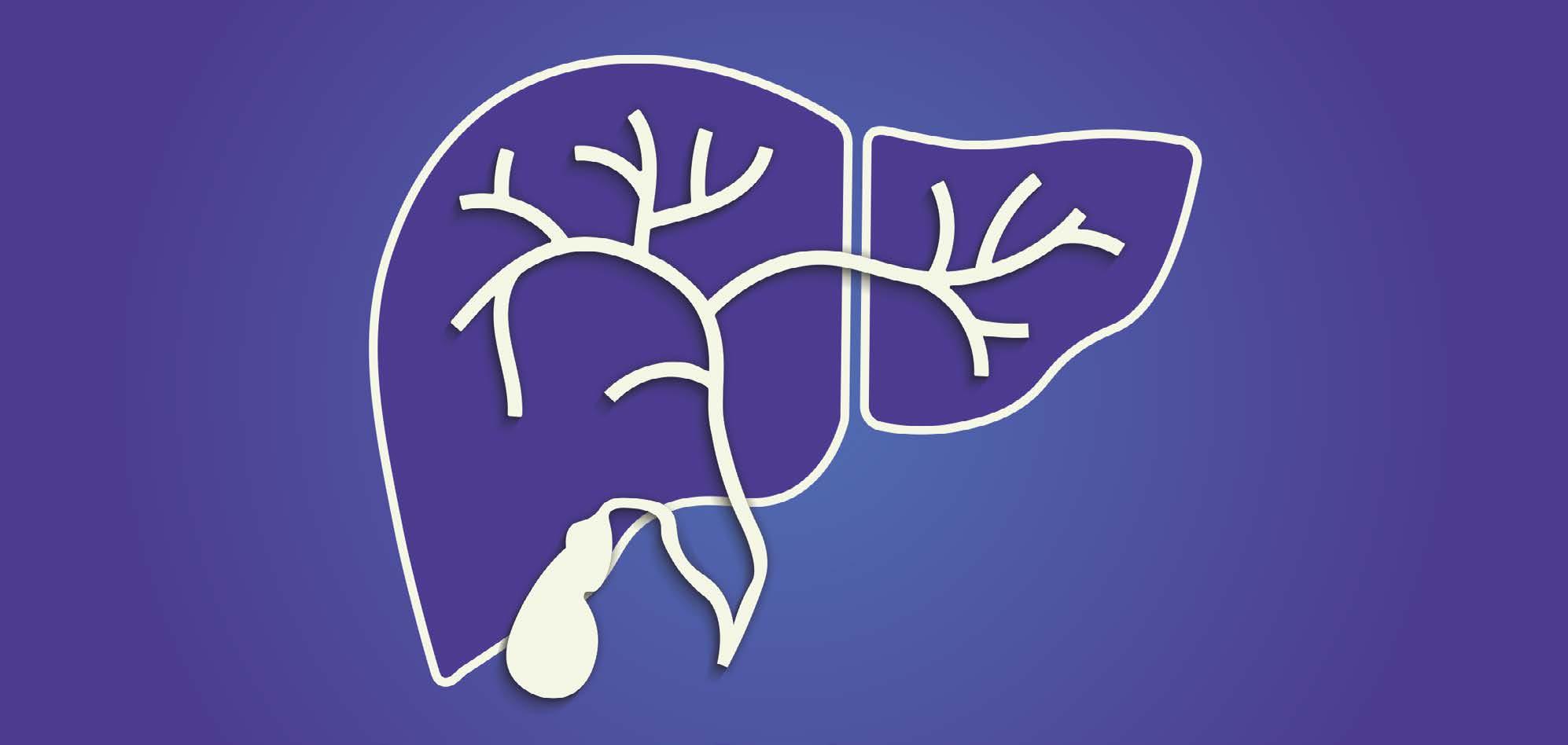
Schematische Darstellung der Leber mit Gallengängen und Gallenblase. Bei PBC sind vor allem die kleinen Gallengänge in der Leber betroffen.
„Die primär biliäre Cholangitis (PBC) ist gut behandelbar!“
Das sagt Univ.-OÄ Priv.-Doz.in DDr.in med. Elisabeth Tatscher von der
Universitätsklinik für Innere Medizin in Graz. Hier klärt die Hepatologin über die seltene Erkrankung auf und gibt Tipps für Betroffene.
PBC ist eine seltene Stoffwechselerkrankung – womit bekommen Betroffene es zu tun?
Die primär biliäre Cholangitis, kurz PBC, ist eine chronische Erkrankung der Leber. Dabei greift das
eigene Immunsystem fälschlicherweise die kleinen Gallengänge in der Leber an. Diese Gallengänge sind wichtig, um die Gallenflüssigkeit aus der Leber abzuleiten, die unter anderem bei der Verdauung von Fetten hilft.
Durch die Entzündung werden die Gallengänge nach und nach geschädigt. Die Galle kann sich dann in der Leber stauen und die Leberzellen schädigen. Über viele Jahre kann dies – unbehandelt – zu einer Vernarbung der Leber
Die Leber ist ein zentrales Stoffwechselorgan. Sie
• verarbeitet Nährstoffe aus dem Darm,
• baut schädliche Stoffe ab (Entgiftung),
• speichert Energie, etwa in Form von Glykogen,
• stellt wichtige Bluteiweiße her.
Zusätzlich produziert die Leber kontinuierlich Galle. Diese Flüssigkeit wird über feine Gallengänge aus der Leber abgeleitet. Wenn keine Verdauung stattfindet, wird die Galle in der Gallenblase gespeichert und dort konzentriert. Bei der Nahrungsaufnahme – besonders nach fettreichen Mahlzeiten – wird die Galle über die Gallengänge in den Dünndarm abgegeben. Dort unterstützt sie die Verdauung und Aufnahme von Fetten sowie der fettlöslichen Vitamine A, D, E und K.
Warum das bei PBC wichtig ist
Bei der Primär biliären Cholangitis (PBC) sind vor allem die kleinen Gallengänge in der Leber entzündet. Dadurch kann der Abfluss der Galle gestört sein, was die Leberzellen schädigen und den Krankheitsverlauf beeinflussen kann. Wichtig zu wissen: Die Gallenblase selbst ist bei PBC nicht die Ursache der Erkrankung – betroffen sind ausschließlich die Gallengänge innerhalb der Leber.

Univ.-OÄ Priv.-Doz. in DDr. in Elisabeth Tatscher Fachärztin für Innere Medizin mit dem Schwerpunkt Hepatologie, Klinische Abteilung für Gastroenterologie und Hepatologie, Universitätsklinik für Innere Medizin, Medizinische Universität Graz
(Leberfibrose) und in schweren Fällen zu einer Leberzirrhose führen.
Die PBC gilt als seltene Erkrankung. Zwei bis 40 pro 100.000 Menschen sind betroffen, wobei die Häufigkeit regional schwankt. Bis zum Alter von 65 Jahren sind 90 Prozent der Betroffenen weiblich. Ab 65 liegt das Verhältnis zwischen Frauen und Männern bei vier zu eins. Zumeist sind die Patient:innen bei der Diagnose 45 bis 65 Jahre alt. Daten zeigen, dass die Erkrankung umso aggressiver voranschreitet, je jünger die Patient:innen in der genannten Altersspanne sind.
Wie zeigt sich die PBC, und wie stellen Sie die Diagnose?
Anfangs spüren Betroffene meist nichts, denn die Leber tut nicht weh. Die Erstdiagnose erfolgt meist als Zufallsbefund wegen anhaltend erhöhter Leberwerte – insbesondere alkalische Phosphatase (ALP) und/oder Gamma-Glutamyltransferase (GGT). Diese müssen labortechnisch, mit bildgebenden Verfahren, zum Beispiel Ultraschall, und gegebenenfalls auch mit einer Gewebeprobe (Biopsie) abgeklärt werden, um andere Ursachen auszuschließen und eine etwaige PBC-Diagnose zu stellen. Bei 90 bis 95 Prozent der PBC-Betroffenen lassen sich im Blut krankheitsspezifische antimitochondriale Antikörper (AMA) nachweisen.
Im weiteren Verlauf können PBC-typische Symptome auftreten: Fatigue, also körperliche und geistige Ermüdung/Erschöpfung, und Juckreiz (Pruritus, vorwiegend an Händen, Füßen und Rücken) kommen bei bis zu 70 Prozent aller Betroffenen vor. Viele Patient:innen leiden auch an trockenen Schleimhäuten, insbesondere der Augen und des Mundes (SiccaSymptomatik). Während Fatigue und Sicca-Syndrom-Symptome unspezifisch sind, sollten behandelnde Mediziner:innen bei Juckreiz in Verbindung mit erhöhten Leberwerten hellhörig werden.
Wie behandeln Sie PBC?
Die Behandlung der PBC ist heute deutlich individualisierter und
orientiert sich am Krankheitsrisiko sowie an den Bedürfnissen der Patient:innen.
Zur Behandlung hat sich seit vielen Jahren eine wasserlösliche, körpereigene Gallensäure bewährt, die gewichtsabhängig dosiert wird. Drei Prozent dieser vergleichsweise milden Gallensäure stecken von Natur aus in der Gallenflüssigkeit, medikamentös erhöhen wir diesen Anteil nötigenfalls auf bis zu 50 Prozent. Auf diese Weise machen wir die Galle weniger „aggressiv“, sodass die ohnehin schon im Zuge der PBC geschädigten Gallengänge geschont werden. Der Gallenfluss –und damit der Stoffwechsel – wird verbessert und der Fortschritt der Erkrankung verlangsamt.
Die PBC ist nicht heilbar, aber gut behandelbar. Mit dieser Behandlung, die lebenslang angewendet werden muss, können wir die Leber lange funktionstüchtig halten. Etwa 70 Prozent der Patient:innen sprechen darauf gut an. Sie überleben nahezu einschränkungs- und vor allem transplantationsfrei.
Bei den verbleibenden 30 Prozent ist das Risiko für einen Fortschritt der PBC höher, insbesondere dann, wenn sie jünger als 45 Jahre alt sind oder bereits an einer Leberfibrose leiden. Für diese Patient:innen stehen inzwischen
zusätzliche therapeutische Möglichkeiten zur Verfügung.
Was raten Sie PBC-Patient:innen zur Bewältigung ihrer Erkrankung?
1. Nehmen Sie Ihre Medikamente wie verschrieben und scheuen Sie sich nicht, au ommende Unverträglichkeiten mit Ihren Hepatolog:innen zu besprechen.
2. Lassen Sie Ihre Leberwerte regelmäßig kontrollieren, insbesondere die alkalische Phosphatase (ALP), die als zentraler Marker für den Krankheitsverlauf gilt.
3. Informieren Sie Ihre Hepatolog:innen gegebenenfalls über ihre Symptome, insbesondere Fatigue, Juckreiz und Sicca-Syndrom, da diese die Lebensqualität erheblich beeinträchtigen. Physis und Psyche können massiv darunter leiden – eine psychologische Begleitung ist mitunter ratsam.
4. Halten Sie sich zur PBC auf dem Laufenden, beispielsweise über die Hepatitis Hilfe Österreich –Derzeit findet in diesem Bereich intensive Forschung statt.
5. Leben Sie lebergesund: Achten Sie auf Ernährung, Bewegung, Schlaf und Stress.
6. Tauschen Sie sich in Selbsthilfegruppen zur Krankheitsbewältigung im Alltag aus.
Ein gesunder Lebensstil kann Ihre Leberfunktion unterstützen und Ihr Wohlbefinden im Alltag steigern. Kleine Veränderungen können schon einen großen Unterschied machen.
Alkoholkonsum
Moderater, vorsichtiger Alkoholkonsum ist für manche Betroffene möglich, sollte aber individuell mit Ihrer Ärztin/Ihrem Arzt abgestimmt werden. Bei fortgeschrittener Lebererkrankung wird empfohlen, auf Alkohol zu verzichten.
Ernährung
Die meisten Menschen mit PBC im Frühstadium können eine gesunde Ernährung ohne Einschränkungen genießen. Wenn Probleme mit der Fettaufnahme auftreten, achten Sie
auf mögliche Vitaminmängel (A, D, E, K) und besprechen Sie dies mit Ihrer Ärztin/Ihrem Arzt.
Bewegung
Körperliche Bewegung kann das allgemeine Energielevel und das Wohlbefinden verbessern.
Mentale Gesundheit
Eine chronische Erkrankung kann psychisch belasten. Wenn Sie sich über längere Zeit niedergeschlagen oder ängstlich fühlen, sprechen Sie mit Ihrer Ärztin/Ihrem Arzt. Selbsthilfegruppen oder Patientenorganisationen bieten Austausch, Rat und Unterstützung.
Soziales Leben und Arbeit
Planen Sie Zeit für Familie, Freunde und Aktivitäten ein, die Ihnen Freude bereiten. Wenn Müdigkeit Ihren Arbeitsalltag erschwert, besprechen Sie mögliche Anpassungen – zum Beispiel flexible Arbeitszeiten oder Pausen.
Mit freundlicher Unterstützung von Ipsen Pharma Austria GmbH
Kristina Sokolovic lebt mit primär biliärer Cholangitis (PBC) und berichtet von ihrem Diagnoseweg, den Herausforderungen und der Kraft, die ihr Wissen und Austausch geben.
Wie begann deine Geschichte mit der primär biliären Cholangitis (PBC)?
Ausgangspunkt war der Tod meines Vaters mit Leberzirrhose: Mir wurde eine Abklärung empfohlen. Da es mir gut ging, habe ich gezögert, und erst auff ällige Leberwerte brachten den Verdacht auf PBC. Diese mögliche Erkrankung wollte ich nicht wahrhaben.
Wie ging es dann weiter?
Erschöpfung und starker Juckreiz wurden zu ständigen Begleitern. 2017 ließ ich mich untersuchen; die Einschätzung schwankte zwischen PBC und PSC. Sogar von einer möglichen Transplantation „in 15 Jahren“ war die Rede. Das musste ich sacken lassen.
Deine Diagnose – wann kam sie und was veränderte sie für dich? PBC wurde im Jahr 2021 bestätigt – endlich hatten meine Symptome einen Namen. Der Juckreiz ließ durch eine medikamentöse Therapie fast vollständig nach, die Müdigkeit schwankt weiterhin.
Wie sieht dein Alltag mit PBC aus?
Als alleinerziehende Konstruktionsingenieurin entlaste ich mich mit einer Vier-Tage-Woche. Bewegung hält mich im Gleichgewicht – Tennis, Radfahren und kurze Dehnroutinen. Beim Essen folge ich einem achtsamen, undogmatischen Ansatz.
Was tun, um der Müdigkeit entgegenzuwirken?
Ich plane Puffer ein, nutze meine

„helleren“ Phasen für Wichtiges und akzeptiere Pausen. Gute Schla ygiene hilft – feste Zeiten, kein Handy, frische Luft. Und wenn nichts geht, verschiebe ich Termine, statt mich zu überlasten.
Gibt es regelmäßige Kontrollen und Therapien?
Einmal jährlich mache ich in der Uniklinik den großen Check, alle drei Monate prüft meine Hausärztin die Blutwerte. So können wir die Therapie frühzeitig anpassen – das gibt mir Sicherheit.
Wie nimmst du die psychische Belastung wahr?




Die Erkrankung bleibt präsent, selbst wenn ich versuche, sie auszublenden. Ich denke sie mit, ohne ihr die Kontrolle zu überlassen. Unterstützung anzunehmen, Grenzen zu setzen und gute Tage bewusst wahrzunehmen – all das hilft.
Was würdest du frisch Diagnostizierten und ihren Angehörigen raten?
Fragen sammeln, Routinen leben, Kontrolle ernst nehmen – so bleibt man handlungsfähig. Angehörige helfen, das Leben aktiv zu gestalten. Wissen und Austausch geben Kraft und eröffnen neue Möglichkeiten.
Wie lebst du heute mit der PBC?
Wichtig ist, dass ich weiß, wie ich mit meiner Erkrankung umgehen muss. Ich habe eine positive Einstellung zu meiner PBC gefunden. Ich mache alles, was mir Spaß macht – und das ist auch mein Lebensmotto. Gleichzeitig brauche ich Erholungsphasen und Zeit zum Durchatmen. Zudem achte ich auf meine Ernährung und ausreichend Bewegung.
Wie können Ärztinnen und Ärzte noch besser unterstützen?
Klarheit, Raum für Fragen und ehrliche Antworten – ohne Schönreden, ohne Angstmachen. Verständliche Sprache und praktische Tipps helfen, den Alltag trotz Krankheit gut zu gestalten.
Mit freundlicher Unterstützung von
Betroffene mit schweren oder seltenen Erkrankungen und ihre Angehörigen brauchen neben medizinischen Informationen auch Raum für Austausch und Orientierung. Genau hier setzt die Initiative „Räume zum Reden“ an – als Plattform, die Wissen bündelt und Perspektiven verbindet.
„Räume zum Reden“ versteht sich als Initiative, in der sowohl Betroffene mit schweren oder seltenen Erkrankungen als auch pflegende Angehörige gesehen, gehört und unterstützt werden. Wer eine Diagnose erhält oder ein betroffenes Familienmitglied im Alltag begleitet, ist täglich mit komplexen Herausforderungen konfrontiert: körperlich, emotional, organisatorisch und häufig auch sozial. Viele Menschen fühlen sich in dieser Situation alleingelassen – obwohl sie enorme Verantwortung tragen und einen unverzichtbaren Beitrag zur Versorgung leisten. Genau hier setzen wir an. Die Initiative gibt dem Engagement der Angehörigen, die kranke Familienmitglieder unterstützen oder pflegen, eine neue Plattform. Gleichzeitig richtet sie sich an Betroffene, die sich verlässliche Informationen, Orientierung und Austausch wünschen. Unser Ziel ist es, beiden Gruppen einen geschützten Raum zu bieten, in dem sie ihre Perspektiven teilen, Verständnis finden und Entlastung erfahren können. Räume zum Reden ist daher mehr als ein Informationsangebot. Es ist eine Plattform für Austausch, ein Ort der Stärkung und ein Beitrag dazu, dass die Bedürfnisse von Betroffenen und pflegenden Angehörigen den Raum bekommen, den sie brauchen und verdienen. Unser Anspruch ist, Informationen nicht nur bereitzustellen, sondern so aufzubereiten, dass sie Menschen im Alltag wirklich weiterhelfen. Dafür nutzen wir unser gesamtes Netzwerk: Ärztinnen und Ärzte, Angehörige und Patient:innen. Die Inhalte unserer Plattform verbinden medizinisches Fachwissen mit alltagsnahen Perspektiven und dem Erfahrungswissen anderer Betroffener und Familien, die ähnliche Wege gegangen sind.
PBC: eine seltene Erkrankung der Leber mit vielfältigen Herausforderungen Die Relevanz solcher Informationsräume wird besonders deutlich bei seltenen Lebererkrankungen wie der primär biliären Cholangitis (PBC). Viele Betroffene warten Jahre auf eine Diagnose – während Müdigkeit, Juckreiz oder unspezifische Schmerzen ihr Leben zunehmend einschränken. PBC ist chronisch, nicht heilbar und dennoch kaum sichtbar. Diese Unsichtbarkeit macht es schwer, im Alltag Verständnis zu finden. Auf raeume-zum-reden.eu finden Patient:innen detaillierte Ressourcen, die ihre eigenen Erfahrungen widerspiegeln. Sie erfahren zum
Beispiel, wie belastend es ist, wenn der Körper sich trotz wieder unauffälliger Laborwerte dennoch weiter erschöpft fühlt und dies im Umfeld oft auf Unverständnis stößt. Hier können sich Betroffene verstanden fühlen und lernen, was hinter Symptomen steckt, die man von außen nicht sieht.
Auf Räume zum Reden finden
Sie:
• verständliche Ratgeber und Wissensartikel
• Tipps zur Stärkung der eigenen Selbstfürsorge
• Mutmacher-Geschichten und Erfahrungsberichte Podcasts, Online-Angebote und


Eine Initiative von Ipsen Jetzt QR-Code scannen und mehr erfahren

Wer täglich verdient selbst
Erfahrungen von Betroffenen, Angehörigen Expert*innen
Praktische Hilfestellungen mit schwerwiegenden, chronischen
Die Initiative „Räume verschafft den pflegender
Mit freundlicher Unterstützung von Roche Austria GmbH
Priv.-Doz.in Dr.in med. univ. Katharina Thom, Kinderfachärztin für Gerinnungserkrankungen und Kardiologin, erklärt, was sie Eltern im Erstgespräch mit auf den Weg gibt, wenn ein Kind die Diagnose der seltenen Erkrankung Hämophilie erhält.

Priv.-Doz. in Dr. in med. univ. Katharina Thom Oberärztin Abteilung für Pädiatrische Kardiologie/Kinderherzzentrum Wien und Gerinnungsambulanz für Kinder und Jugendliche, Spezialistin für Pädiatrische Kardiologie und Gerinnungsstörungen, Universitätsklinik für Kinder- und Jugendheilkunde, Medizinische Universität Wien
Wie bauen Sie ein Erstgespräch auf, wer sollte neben den Eltern am Tisch sitzen und welche Informationen sind essenziell?
Die Neudiagnose „Hämophilie“ belastet Eltern besonders, wenn die Erkrankung in der Familie nicht bekannt war und ihr Kind mit einer akuten Blutung zunächst versorgt werden muss. Bei einer Gerinnungsstörung als Ursache für die Blutung sind dann ausführliche Au lärungsgespräche erforderlich. Die Hämophilie ist eine seltene Gerinnungsstörung, die fast nur Jungs betrifft und bei der Gerinnungsfaktoren im Blut fehlen: Faktor VIII bei der Hämophilie A oder Faktor IX bei der Hämophilie B. Die A-Form ist häufiger mit einem von 5.000 männlichen Neugeborenen als die B-Form mit einem von 25.000 männlichen Neugeborenen. In den ersten Gesprächen nach Diagnosestellung wird erläutert, dass ein solcher Faktormangel meist angeboren ist. Die resultierende Blutungsneigung ist abhängig vom Schweregrad der Hämophilie. Normalerweise haben wir über 60 % Faktor VIII oder IX im Blut. Bei der Hämophilie wird unterschieden zwischen leichter Hämophilie (Faktor VIII oder IX unter fünf Prozent), mittelschwerer (eigener Restfaktor zwei bis fünf Prozent) oder schwerer (weniger als ein Prozent Faktor) Form. Der individuelle Blutungsphänotyp kann jedoch variieren. Während Patienten mit schwerer Hämophilie häufig spontane Blutungen entwickeln, treten bei leichteren Formen Blutungen meist nach Verletzungen oder Operationen auf. Patienten mit einer schweren Hämophilie haben ein höheres Risiko für Blutungen in Gelenke,
Muskeln und Weichteile, aber auch Schleimhautblutungen. Am häufigsten ist eine gesteigerte Hämatomneigung (blaue Flecken), die oft auch zur Abklärung führt. Die Erkrankung verläuft zwar chronisch, ist aber sehr gut therapierbar. Die klassische Behandlung der Hämophilie erfolgt durch den Ersatz des fehlenden Gerinnungsfaktors über die Vene. Bei leichter oder mittelschwerer Hämophilie wird der Faktor nur bei Bedarf, beispielsweise zum Stillen einer Blutung und vor einer OP, gegeben. Bei der schweren Hämophilie ist eine Prophylaxe nötig – über die Vene oder mit neuen Non-Faktor-Therapien subkutan (unter die Haut). Das Ziel der Prophylaxe ist vor allem, Gelenkblutungen und chronische Gelenkschäden zu verhindern sowie schwere innere Blutungen, einschließlich seltener, aber potenziell lebensbedrohlicher Hirnblutungen, zu vermeiden. Nach Erstdiagnose einer Gerinnungsstörung wie der Hämophilie sehen wir die Familien und gegebenenfalls weitere Bezugspersonen zu einem zeitnahen weiteren Gespräch in der Kindergerinnungsambulanz. Hierbei wird auch erläutert, wie die Eltern in Blutungssituationen, wie unklaren Schwellungen von Gelenken oder Muskeln, reagieren sollten und an wen sie sich wenden können für eine rasche und fachkundige Behandlung.
Seltene Erkrankungen wie Hämophilie werden oft erst spät erkannt. Wie schätzen Sie die Sensibilisierung der Erstversorgenden in Österreich ein?
Ich erwarte bei einer seltenen Erkrankung nicht, dass sich alle
1 https://bluter.at/wp/wp-content/uploads/2021/10/haemophilie_leitlinien.pdf
2 https://bluter.at/wp/arbeitsschwerpunkte-projekte
damit auskennen. Die Praxis zeigt jedoch, dass viele Kinderärztinnen und -ärzte ein gutes Bewusstsein für eine gesteigerte Blutungsneigung haben und entsprechende Tests veranlassen beziehungsweise Patienten an uns überweisen. Verzögerungen bei der Diagnosestellung entstehen am häufigsten in Situationen, wo die Hämophilie in der Familie nicht bekannt war.
Wie ist Österreich im Bereich Hämophilie aufgestellt – sowohl in Bezug auf die Zahl der Spezialist:innen als auch auf die interdisziplinäre Betreuung? Die interdisziplinäre Betreuung von Patienten mit Hämophilie und anderen Gerinnungsstörungen ist hierzulande gut. Gerade in Spitälern stehen Teams bereit, die Expertisen aus Pflege, Physiotherapie, Gerinnungserkrankungen und auch Psychotherapie vereinen.
Wir Gerinnungsexpert:innen sind landesweit gut vernetzt und haben erst kürzlich unsere gemeinsame „Leitlinie zur Hämophiliebehandlung in Österreich“1 aktualisiert.
Auch die Österreichische Hämophilie Gesellschaft (ÖHG) ist sehr aktiv: Sie verbindet alle Behandelnden und Betroffenen und bietet Raum für Information, Austausch und Beratung. Hervorheben möchte ich an dieser Stelle das „Sommercamp“2 – ein seit mehr als 50 Jahren jährlich stattfindendes Camp für Kinder und Jugendliche mit Blutgerinnungsstörungen, das mein Kollege Ao. Univ.-Prof. Dr. med. univ. Christoph Male-Dressler, DGKP Eva Wissmann und ich persönlich betreuen.
Mit freundlicher Unterstützung von
Roche Austria GmbH

Antonia und ihr Sohn Leo beim Wintertreffen der ÖHG
Antonia Kern ist Vorstandsmitglied der Österreichischen Hämophilie Gesellschaft (ÖHG) und Mutter eines Sohnes, der mit Hämophilie geboren wurde. Im Interview berichtet sie über die Bedeutung ausführlicher Diagnosegespräche sowie des Austauschs mit anderen Betroffenen.
Wie kam es bei Ihrem Sohn zur Diagnose?
Leo war da noch nicht einmal ein Jahr alt und hatte blaue Flecken an ungewöhnlichen Stellen. Ich war beim Hautarzt und habe ihn gebeten, sich das anzusehen. Er hat mir dann geraten, die Gerinnung testen zu lassen, was wir dann auch gemacht haben. Dabei wurde schnell klar, dass mit der Gerinnungszeit etwas nicht in Ordnung ist. Dass es sich um eine Spontanmutation handelt und ich Überträgerin bin, stellte sich jedoch erst später heraus, nachdem sich meine Mutter testen ließ und das Ergebnis negativ war. Das war eine einschneidende Erfahrung.
Wie haben Sie die Erstdiagnose erlebt?
Wir wurden lediglich telefonisch verständigt. Es gab da keine Möglichkeit, das zu besprechen und Fragen zu stellen, um es irgendwie einordnen zu können. Ein echtes Diagnosegespräch fand erst zwei Wochen später im Wiener AKH statt. Die Wartezeit war von großer Unsicherheit geprägt. Ich habe die Zeit vor allem damit verbracht, im Internet nach Informationen zu suchen. Natürlich machte ich mir auch Sorgen, ob Leo normal aufwachsen und wie seine große Schwester in die Krabbelstube und den Kindergarten gehen kann. Das Erstgespräch und die Betreuung in der Gerinnungsambulanz im AKH
waren dann aber wirklich super. Auch die ÖHG hat uns in dieser ersten Phase sehr gut unterstützt: Ich habe rasch einen Kontakt zu einer anderen Mutter eines an Hämophilie erkrankten Kindes bekommen und mich sofort mit ihr getroffen. Das hat uns in dieser Situation ein bisschen gerettet.
Wie gehen Sie mit der Erkrankung im Alltag um?
Das erste Jahr mit den vielen Krankenhausbesuchen war sicherlich die schlimmste Phase – sowohl für Leo als auch für uns als Familie. Die regelmäßigen Spritzen waren anfangs sehr herausfordernd. Mit der Zeit haben wir eine Methode gefunden, die gut in unseren Alltag passt, sodass die Behandlung zu Hause inzwischen problemlos gelingt. Wir haben aber auch das Glück, dass Leo trotz einer schweren Verlaufsform praktisch keine Blutungsneigung hat. Wie sehr man achtgeben muss, ist natürlich von Kind zu Kind stark unterschiedlich, aber ich habe nie das Gefühl, dass bei Leo etwas fundamental anders wäre als bei anderen Kindern. Unser soziales Umfeld weiß Bescheid, und wenn nicht, dann erkläre ich, was sie beachten müssen. Wir machen da keine große Sache daraus. Leo ist mittlerweile sieben Jahre alt und geht selbst offen damit um. Sollte er einmal einen Unfall haben, dann gehe ich davon aus, dass er wie
jedes andere Kind schnell ins Krankenhaus kommt. Ich habe sichergestellt, dass sein Präparat dort immer vorrätig ist, damit er entsprechend versorgt werden kann. Bis auf die subkutane Behandlung alle zwei Wochen unterscheidet sich sein Alltag damit kaum von jenem anderer Kinder.
Sie haben die Informationssuche angesprochen. Wie handhaben Sie das?
In der ersten Zeit habe ich aufmerksam bei einer FacebookGruppe in Deutschland mitgelesen und viel über neue Medikamente mitgenommen. Informationen aus dem Internet sollte man aber immer mit den behandelnden Ärzt:innen besprechen. Das Internet ist für mich mittlerweile kaum noch relevant. Die ÖHG bietet mir alle Informationen, die ich brauche – sei es durch Veranstaltungen wie die jährliche Generalversammlung, Familienausflüge oder Sportcamps, durch Vorträge von Ärzt:innen, über die Website bluter.at oder durch Auskünfte am Telefon, per E-Mail oder in unserer Mütter-WhatsAppGruppe. Dort findet sich auf jede Frage rasch eine Antwort. Auch die Vernetzung mit anderen Betroffenen in der Region ist eine super Sache. Wenn mal will, kann man sich natürlich auch selbst einbringen – etwas, das mir große Freude macht.

Antonia Kern Vorstandsmitglied der Österreichischen Hämophilie Gesellschaft, Mutter eines betroffenen Sohnes
Mit freundlicher Unterstützung von Servier Austria GmbH
Daniel Eßletzbichler (37) lebt mit einem seltenen Gehirntumor (diffuses Gliom/Astrozytom). Gemeinsam mit seiner Partnerin Edda Pascher (46) berichtet er vom Weg zur Diagnose, von Rückschlägen und neuen Perspektiven.
Daniel, wie kam es zu deiner Diagnose?
Daniel: Im Sommer 2016 hatte ich aus dem Nichts einen Krampfanfall. Damals wurde mir erklärt, es handle sich vermutlich um eine angeborene Zyste im Kopf, weshalb ich dem Vorfall wenig Bedeutung beimaß. Anfang 2021 bemerkte ich jedoch sprachliche Aussetzer, Tinnitus und Schwindel. Ich schob das auf den Stress, weil wir mit unserem veganen Bio-Lebensmittelgeschäft während der Pandemie stark gefordert waren.
Edda: Im Juni 2021 reagierte
Daniel plötzlich nicht mehr auf meine Ansprache, ich wählte sofort den Notruf.
Daniel: Die Bildgebung in der Notaufnahme zeigte eine große Läsion im rechten Schläfenlappen, halb so groß wie meine Faust, durch deren Druck die Mittellinie meines Gehirns lebensbedrohlich verschoben war. Sofort wurde eine OP geplant.
Wie verlief die Operation?
Daniel: Ein Großteil des Tumors konnte entfernt werden, allerdings kam es zu schweren Komplikationen. Ich hatte eine Nahtoderfahrung, danach lag ich drei Wochen im künstlichen Koma. Nach dem Aufwachen musste ich vieles neu lernen: schlucken, essen, aufstehen, gehen ... Lange konnte ich mich nur mithilfe eines Rollators fortbewegen. Es folgten Chemotherapie und Bestrahlung.
Edda: Diese Zeit war für uns beide sehr belastend. Mehrmals wurde mir während Daniels Koma gesagt, er würde die Nacht nicht überleben oder schwere Schäden davontragen.
Doch du hast es geschafft, Daniel!
Daniel: Mit viel Willenskraft und intensiver Rehabilitation schaffte ich es, wieder ins Leben und sogar in unser Geschäft zurückzukehren.
Doch der Alltag blieb ein ständiger Balanceakt, weil Fatigue, Konzentrationsschwierigkeiten und eine eingeschränkte Merkfähigkeit
weiterhin spürbar waren. Rückblickend hätte ich früher Unterstützungsmöglichkeiten wie Rehageld oder Erwerbsunfähigkeitspension in Anspruch nehmen sollen. Fehlende Beratung und widersprüchliche Informationen haben mich jedoch lange davon abgehalten.
Edda: Fehldiagnosen, unzureichende Au lärung und fehlende Unterstützung sind für viele Betroffene eine große Belastung.
Daniel: Auch wir wurden bei der Diagnose nicht entsprechend aufgeklärt. Informationen zu IDHmutierten Astrozytomen waren schwer zugänglich und sie wurden im Internet oft mit dem aggressiveren Glioblastom verwechselt, was zusätzlich verunsichert hat.
Wie war der weitere Verlauf?
Daniel: Regelmäßige KontrollMRTs zeigten über mehrere Jahre stabile Befunde, sodass ich glaubte, die Erkrankung überwunden zu haben. Anfang 2025 bemerkte ein Neurochirurg durch Zufall, dass der letzte Befund falsch war. Hätte er das nicht bemerkt, wäre ich heute tot, denn der Krebs war in einem aggressiveren Grad zurückgekommen. Es folgten eine weitere OP und erneute Chemotherapie. Unser Geschäft mussten wir daher nach fast zehn Jahren schließen.
Wie geht es dir heute?
Daniel: Ich bin erwerbsunfähig, befinde mich weiterhin in Behandlung und versuche, aktiv zu bleiben und meine körperliche Fitness zu erhalten – bei einer onkologischen

Behandlung ein sehr wichtiger Faktor. Ich konnte viele Hobbys wieder aufnehmen, wie Fotografie, Wetterbeobachtung und Musik. Während meiner Spitalsaufenthalte lernte ich Musiktherapie kennen und habe begonnen, Handpan zu spielen. Diese Aktivitäten helfen mir bis heute sehr.
Edda, welche Rolle hast du seit Daniels Erkrankung übernommen – damals und heute?
Edda: Da wir beide selbstständig waren, konnte ich mir viel Zeit nehmen, um Daniel während der Krankenhausaufenthalte zu begleiten. Das bedeutete allerdings einen erheblichen Verdienstausfall. Auch heute begleite ich Daniel zu medizinischen Terminen und Therapien und bin im Alltag möglichst in seiner Nähe, um im Notfall sofort reagieren zu können.
Was ratet ihr anderen Betroffenen und Angehörigen?
Daniel: Informiert euch und lasst nicht locker! Wichtig ist auch Austausch mit anderen Betroffenen, deshalb engagiere ich mich im Au au einer Patient:innenvernetzung für Gliom-Betroffene. Mein Rat: Selbstbestimmt bleiben, den Rückhalt in Familie und Partnerschaft spüren, sich erreichbare Ziele setzen und den Blick auf das richten, was weiterhin möglich ist. Tägliche Bewegung, frische Luft und ein achtsamer Umgang mit den eigenen Gefühlen helfen, Lebensqualität zu erhalten und hoffnungsvoll nach vorne zu schauen.
Daniel Eßletzbichler und Edda Pascher

Leiterin der Neuroonkologischen Ambulanz, Universitätsklinik für Neurologie, Medizinische Universität Graz
Dr.in med. Tadeja Urbanic Purkart, Leiterin der Neuroonkologischen Ambulanz der Medizinischen Universität Graz, erklärt, worauf es bei der Diagnose und Behandlung der seltenen Gehirntumoren ankommt.
Was ist ein diffuses Gliom, und was bedeutet die Diagnose für Betroffene?
Gliome sind eine heterogene Gruppe primärer Hirntumoren, die aus Gliazellen, den Stützzellen des Gehirns, oder deren Vorläuferzellen hervorgehen. Man unterscheidet umschriebene, klar abgegrenzte Gliome, die überwiegend im Kindesalter auftreten und häufig vollständig operativ entfernt werden können, von den sogenannten diffusen Gliomen.
Zu den diffusen Gliomen des Erwachsenenalters zählen Astrozytome, Oligodendrogliome und Glioblastome. Sie wachsen infiltrativ in das umliegende Hirngewebe ein, lassen sich daher meist nicht vollständig entfernen und begrenzen die Lebenszeit in Abhängigkeit vom Grad der Entartung. Die Diagnose diffuser Gliome mit initial langsamerem Wachstum, sogenannte IDH-mutierte Grad-2-Gliome, ist für die typischerweise 30 bis 45 Jahre jungen Erwachsenen, die mitten im Leben stehen, immer ein Schock.
Wie macht sich ein diffuses
Gliom erstmals bemerkbar?
Meist kommen die Patient:innen wegen eines epileptischen Anfalls in die Notaufnahme. Die Epilepsie ist jedoch nicht nur das typische Erstsymptom, sondern auch ein Leitsymptom: 60 bis 70 Prozent der Patient:innen mit einem primär langsam wachsenden diffusen Gliom leiden daran. Die Epilepsie beeinträchtigt sie im Alltagsleben, im Denken und im Wohlbefinden. Viele haben Angst vor Anfällen und bleiben vorsichtshalber zu Hause –fern von sozialen Kontakten.
Andere Symptome können Persönlichkeitsveränderungen oder Verhaltensstörungen, Probleme mit Aufmerksamkeit und Konzentration, verminderte Belastbarkeit, Lähmungen, Sehstörungen, Schwierigkeiten beim Sprechen oder beim Sprachverständnis sein.
Wie wird ein diffuses Gliom diagnostiziert, und was ist die Herausforderung dabei? Wer nach einem epileptischen Anfall in die Notaufnahme kommt, erhält in der Regel zunächst eine Computertomografie (CT) des Gehirns. Damit lassen sich akute Ursachen wie eine Hirnblutung, ein größerer Schlaganfall oder eine ausgeprägte Hirnschwellung rasch erkennen oder ausschließen. Diffuse Gliome sind im CT jedoch häufig nur unscharf abgrenzbar oder in frühen Stadien nicht eindeutig zu erkennen.
Der Goldstandard in der Bildgebung ist daher die Magnetresonanztomografie (MRT). Erfahrene Neuroradiolog:innen können anhand der Bildmorphologie eine Verdachtsdiagnose stellen und Hinweise auf den wahrscheinlichen Tumortyp und das biologische Verhalten ableiten. Eine definitive Diagnose und molekulare Einordnung gelingt jedoch nur durch die histomolekulare Untersuchung von Tumorgewebe, das im Rahmen einer Biopsie oder Operation gewonnen wird. Wird ein diffuses Gliom nicht erkannt oder in seinem biologischen Verhalten unterschätzt, kann dies fatal sein. Bei einem Verdacht sollte daher zeitnah eine spezialisierte neuroonkologische Abklärung erfolgen.
Wie behandeln Sie diffuse Gliome?
Jedes diffuse Gliom verkürzt das Leben. Ziele unserer Behandlung sind deshalb, dass die Betroffenen möglichst keine epileptischen Anfälle haben, kognitiv lange funktionsfähig und kritikfähig sind und ihre Lebensqualität lange erhalten bleibt. Meist werden bei schneller wachsenden Tumoren OP, Chemotherapie und Strahlentherapie kombiniert. Studien zeigen, dass gerade die Kombi der beiden Letztgenannten das progressionsfreie ebenso wie das Gesamtüberleben signifikant verlängert.
Epilepsie ist ein häufiges Symptom beim diffusen Gliom. Der Internationale Epilepsie-Tag am 9. Februar macht jedes Jahr auf das Thema aufmerksam.
Zwei Drittel der Patient:innen mit diffusem Gliom und Epilepsie sprechen auf eine anfallssuppressive Therapie an. Dahinter steckt der Zusammenhang, dass gerade langsam wachsende diffuse Gliome auch das epileptische Geschehen fördern. Gliom-Betroffene, die anfallsfrei sind, dürfen unter Einhaltung der gesetzlichen Rahmenbedingungen auch Auto fahren. Bestenfalls entscheidet ein interdisziplinäres Team gemeinsam mit den Patient:innen über die Therapie. Solche Tumorboards mit Expert:innen verschiedenster medizinischer Bereiche finden sich österreichweit. Wichtig ist dabei der offene Austausch zwischen den Mediziner:innen und Betroffenen, denn für Letztere gilt es, das Beste aus der Zeit zu machen, die ihnen bleibt. Jeder Therapiepfad ist deshalb ganz individuell.
Unterstützt durch Acadia Pharmaceuticals GmbH Unterstützt
Dr.in med. Fiona Zeiner ist Oberärztin an der Kinderklinik Innsbruck. Im Interview stellt die Kinderneurologin das Rett-Syndrom vor und erklärt, worauf es bei der Diagnose der seltenen Entwicklungsstörung ankommt.

OÄ Dr. in med. Fiona Zeiner Fachärztin für Kinder- und Jugendmedizin mit Schwerpunkt Neuropädiatrie und Entwicklungsneurologie, Universitätsklinik für Pädiatrie I, Medizinische Universität Innsbruck
Was ist das Rett-Syndrom?
Das ist eine seltene, genetisch bedingte Entwicklungsstörung. Die Häufigkeit wird mit etwa einem von 10.000 bis 15.000 Mädchen angegeben, was in Österreich rund drei bis fünf Neuerkrankungen pro Jahr entspricht. Sie betrifft fast ausschließlich Mädchen.
Wie zeigt sich die seltene Erkrankung?
Die betroffenen Kinder entwickeln sich zunächst unauff ällig. Mit sechs bis 18 Monaten kommt es zu einem Entwicklungsstopp. Statt sich weiterzuentwickeln, verlieren die Kinder bereits erworbene sprachliche und motorische Fähigkeiten. Diese Rückschritte in der Entwicklung sind für Kinderärztinnen und -ärzte ein Alarmsignal. Auch das Kopfwachstum ist anfangs normal und wird dann langsamer als bei gesunden Kindern.
Charakteristisch für Kinder mit Rett-Syndrom ist das Entwickeln von typischen Handbewegungen wie Waschbewegungen und im Verlauf der Verlust einer sinnvollen Handfunktion. Bei mildem Verlauf ist dieses Symptom leicht ausgeprägt, aber doch vorhanden. Es braucht Erfahrung, um zu erkennen, welches Kind früh weiter abgeklärt werden muss. Dann wird im Rahmen einer genetischen Untersuchung die Diagnose gestellt.
Wie verläuft die Erkrankung?
Viele Kinder mit Rett-Syndrom lernen gehen, wobei sie sich nicht so geschmeidig bewegen wie gesunde Gleichaltrige, sondern auffallend breitbeinig unterwegs sind. Manche lernen nie zu sprechen, mache verlernen zu sprechen, andere
sprechen einzelne Wörter oder in einfachen Sätzen.
Mit der Zeit stellen sich Probleme mit der Ernährung und Verdauung ein. Die Kinder bleiben meist kleiner und leichter als Gleichaltrige. Im Jugendalter besteht oft eine schwere Schlafstörung, die für die Familie eine hohe Belastung darstellt.
Acht von zehn Personen mit Rett-Syndrom entwickeln eine Epilepsie.
Die Lebenserwartung der Betroffenen liegt im Schnitt bei 40 Jahren, hängt aber vom Schweregrad des Rett-Syndroms und etwaigen Begleiterkrankungen ab.
Warum ist eine frühe Diagnose wichtig?
Ich erlebe, dass die Diagnose viele Eltern entlastet, weil sie eine Antwort auf die sie quälende Frage „Was ist los mit meinem Kind?“ erhalten. Die Krankheit ist unheilbar. Eine frühe Diagnose führt nicht zu einer besseren Behandlung. Schon vor der Diagnose werden Kinder mit einer Entwicklungsstörung ganzheitlich gesehen und gefördert, um ihre Fähigkeiten zu verbessern. Der erste Schritt ist hier fast immer die Frühförderung. Bei der Frühförderung kommen die Fachkräfte ins Haus. Sie fördern das erkrankte Kind und unterstützen zugleich die Familien beim Verarbeiten der Diagnose. Individuell werden Logopädie, Ergotherapie oder Physiotherapie in das Förderkonzept eingebaut. Wir schauen immer, was das Kind kann, und zielen mit der Therapie auf Lebensqualität und Teilhabe am Familienleben und am gesellschaftlichen Leben ab. Ganz wichtig ist mir, zu versichern, dass Personen mit Rett-Syndrom viel mehr am
sozialen Austausch teilhaben, als sie zeigen können. Daher ist es wichtig, Kindern, die kaum sprechen können, eine andere Möglichkeit zu geben, sich auszudrücken. Die Unterstützte Kommunikation hilft durch Karten mit Bildern oder Symbolen oder mit einem einfachen Sprachcomputer. Personen mit Rett-Syndrom verstehen oft mehr, als sie sagen können.
Was raten Sie Eltern, die sich wegen einer Entwicklungsverzögerung bei ihren Kindern sorgen?
Eltern kennen ihre Kinder in der Regel am besten. Wenn ihnen an ihren Kindern etwas auff ällt und sie sich Sorgen machen, sollten sie das immer abklären lassen – am besten von ihrem Kinderarzt oder ihrer Kinderärztin.
Der genetische Nachweis des Rett-Syndroms ist eindeutig. Dank der Fortschritte in der Genetik erhalten Familien mit Kindern mit einer anderen Entwicklungsstörung ebenfalls immer früher eine Antwort auf die Frage, warum sich ihr Kind auff ällig entwickelt.
Und welchen Rat geben Sie zur bestmöglichen Bewältigung der Erkrankung?
Kinder mit Rett-Syndrom sind höchst pflegebedürftig. Das belastet die Familien. Ich erlebe dennoch viel Lebensfreude in den Familien. Vielen hilft der Austausch mit anderen betroffenen Familien.
Leider warten heute viele Eltern mit Kindern mit Entwicklungsstörungen monatelang auf Frühförderung oder auf die empfohlene Therapie. Ich wünsche mir für die betroffenen Familien, dass sich diese Situation ändert, da die Kinder Zeit verlieren.
Unterstützt durch Acadia Pharmaceuticals GmbH
Sophie leidet am Rett-Syndrom. Ihre Eltern Johann, der auch Präsident des Selbsthilfevereins Österreichische RettSyndrom Gesellschaft (ÖRSG) ist, und Sonja Lang berichten, was die Diagnose bedeutet und wie die Familie, zu der auch Sophies ältere Schwester Laura gehört, ihren Alltag mit der seltenen Erkrankung bewältigt.
Wie kam es zu Sophies Diagnose?
Sonja: Sophie war in allem später dran als ihre große Schwester. Doch das beunruhigte uns anfangs nicht. Sie nimmt sich ihre Zeit, dachten wir. Als sie mit acht Monaten ihr Köpfchen nicht hob, sprachen wir das in der Kinderarztpraxis an. Sophie bekam Physiotherapie. Sie lernte krabbeln, hantierte mit Löffel und Gabel. Mit 13 Monaten bemerkten wir weniger Fortschritte in Sophies Entwicklung.
Johann: Sie krabbelte weniger, zog sich nicht hoch, stellte sich nicht auf.
Sonja: Eine Kollegin erzählte mir von einem Mädchen, das das Rett-Syndrom hatte – es gab Parallelen zu Sophie. Ein Gentest bestätigte den Verdacht.
Wie nahmen Sie die Diagnose auf?
Johann: Unsere Welt brach zusammen. Sophie hat einen unheilbaren Gendefekt. Sie wird nie selbstständig leben können.
Wie sieht der Alltag mit Sophie aus, und was macht Ihre Tochter besonders gerne?
Sonja: Sophie braucht immer helfende Hände. Ihre Pflege wird anstrengender, je älter sie wird. Denn gleichwohl sie mit 17 nur 25 Kilogramm wiegt, weil sie keine Muskeln hat, und auch kleiner als gesunde Gleichaltrige ist, kostet das ständige Heben und Tragen Kraft. Ohne Muskeltonus ist sie immer wie ein sehr schläfriges Kind in unseren Armen. Eltern wissen, was ich meine.
Johann: Wenn wir mit Sophie zusammen sind, führen wir alle Selbstgespräche. Das ist okay, solange wir sehen und spüren, dass es Sophie gut geht. Ihr Lächeln ist unwiderstehlich.
Sonja: Wir wissen, dass Sophie mehr versteht, als sie uns mitteilen

kann. Das ist besonders schwer zu ertragen, wenn es Sophie mal nicht gut geht und sie uns nicht sagen kann, was ihr fehlt.
Johann: Sophie liebt Schwimmen, aber nur, wenn das Wasser 30 Grad und wärmer ist. Kürzlich war sie im Kino und hatte großen Spaß mit Pumuckl. Ihre Pumuckl-Puppe lässt sie seitdem nicht aus den Augen.
Was fordert Sie als pflegende Eltern besonders heraus?
Sonja: Sophie taktet unser Leben. Wir brauchen für Alltägliches viel Zeit, insbesondere für die Mahlzeiten. Tags geht sie in eine Sonderschule und absolviert einige ihrer Therapien außer Haus. Sobald sie wieder daheim ist, ist einer von uns beiden für sie da. Das machen wir, solange wir können. Krankheitsbedingt schläft Sophie nachts schlecht. Morgens um halb drei, vier beginnt ihr Tag. Das ist besonders anstrengend.
Johann: Das Rett-Syndrom fordert die betroffene Familie oft auch finanziell heraus. Es gibt inzwischen moderne Hilfsmittel, die die Mobilität, Kommunikation und Teilhabe der Erkrankten am Alltag verbessern, beispielsweise Augensteuerungsgeräte. Doch die müssen sich die Familien leisten können – der Staat unterstützt das deutlich weniger als beispielsweise
den Heizungstausch.
Wie unterstützt die ÖRSG betroffene Familien?
Johann: Der Selbsthilfeverein, dessen Arbeit größtenteils auf Spenden basiert, ist die Anlaufstelle von betroffenen Familien für betroffene Familien. Wir bieten seit 1993 Information, Au lärung, Austausch und Vernetzung untereinander und mit Expert:innen. Aktuell sind in der Österreichischen Rett-Syndrom Gesellschaft rund 120 Kinder mit Rett-Syndrom registriert, darunter auch zwei Buben. Manchen Eltern frisch diagnostizierter Kleinkinder fällt es schwer, in den Austausch mit Eltern älterer Betroffener zu treten. Es schmerzt, die Zukunft seines voll pflegebedürftigen Kindes direkt vor Augen geführt zu bekommen. Doch der Austausch ist sehr wertvoll für alle und hilft beim Bewältigen des Alltags mit Rett-Syndrom in der Familie.
Johann Lang und Sonja Lang, Eltern der betroffenen
Sophie
Weitere Informationen unter: rett-syndrom.at
Sie möchten die ÖRSG mit einer Spende unterstützen? Hier geht’s zum Spendenkonto:

Österreichische Rett-Syndrom Gesellschaft
Bank Austria
IBAN: AT60 1200 0100 2914 9969
BIC: BKAUATWW

Mit freundlicher Unterstützung von Biogen Austria GmbH und Roche Austria GmbH
Bei „UpDate Muskelforschung 2026“ diskutierten Expert:innen aus Medizin, Wissenschaft und Selbsthilfe in Wien über ein Thema, das noch vor wenigen Jahren kaum denkbar gewesen wäre: „Kinderwunsch, Schwangerschaft und Elternschaft mit einer Muskelerkrankung“. Die Tagung machte deutlich, wie sehr sich die Perspektiven für Betroffene von neuromuskulären Erkrankungen in den letzten Jahren verändert haben.
Mit Therapien, die seit wenigen Jahren in Österreich für die häufigsten Muskelerkrankungen wie Muskeldystrophie Duchenne und Spinale Muskelatrophie (SMA) verfügbar sind, ist es vielen Patient:innen möglich geworden, das Erwachsenenalter zu erreichen – und somit Lebensentwürfe zu entwickeln, die Ausbildung, Beruf und Familienplanung einschließen.
Weiterentwickelte Behandlungsmöglichkeiten im Erwachsenenalter Früher bestand teilweise Skepsis hinsichtlich der therapeutischen
Möglichkeiten für Erwachsene mit 5q-assoziierter SMA. Inzwischen zeigt sich jedoch, dass sich die Behandlungsoptionen auch im Erwachsenenalter positiv auf den Krankheitsverlauf auswirken können. Für viele Patient:innen mit SMA bedeutet dies einen Zugewinn an Selbstständigkeit oder zumindest deren Erhalt – eine zentrale Voraussetzung für die Übernahme elterlicher Verantwortung.
Junge Menschen mit einer Muskelerkrankung: Familienplanung wird Teil der Lebensrealität Die Tagung der Österreichischen Muskelforschung zeigte, dass
medizinische Fragen zu Kinderwunsch und Elternschaft heute differenziert und evidenzbasiert diskutiert werden können. Gleichzeitig wurde betont, wie wichtig interdisziplinäre Betreuung, psychosoziale Unterstützung und individuelle Beratung sind. Dank therapeutischer Fortschritte, wachsender Erfahrung und solider Real-World-Daten entsteht ein neues Kapitel in der neuromuskulären Medizin – eines, das den Betroffenen nicht nur Lebenszeit, sondern auch Lebensqualität schenkt und Lebensplanung erlaubt.

die Videoenzyklopädie für neuromuskuläre Erkrankungen auf muskelforschung.at

Die Seltenen Erkrankungen brauchen starke Netzwerke – über Berufsgruppen, Regionen und Ländergrenzen hinweg. Die 2. Tagung für Seltene Erkrankungen der DACH-Region (DACH-SE) bringt die beteiligten Akteure zusammen.
Zwei Tage lang widmen wir uns aktuellen wissenschaftlichen Erkenntnissen, innovativen Konzepten und praxisnahen Impulsen. Das Programm umfasst Übersichtsvorträge, Kurzpräsentationen und Diskussionsformate - mit viel Raum für Vernetzung und fachlichen Austausch.
Themenschwerpunkte u. a.:
• Seltene Erkrankungen in der Praxis – Was bringt uns voran
• Transition – Best Practice
• Digitale Werkzeuge für Diagnostik und Versorgung
• Genomische Diagnostik in der DACH-Region – Erfolge und Herausforderungen
• Neue Erkenntnisse zu genetischen Mechanismen jenseits der DNA-Sequenz
• Innovative Therapieansätze und Versorgungskonzepte
Die Tagung richtet sich an alle, die an der Versorgung von Patient:innen mit seltenen Erkrankungen beteiligt sind - ärztliche und nichtärztliche Fachkräfte. Ebenso freuen wir uns auf die Teilnahme von Patientinnen und Patienten, Selbsthilfeorganisationen und deren Dachverbände (z. B. ACHSE e. V.).
Gemeinsam stärken wir ein multiprofessionelles, länderübergreifendes Netzwerk – für bessere Diagnosen, bessere Versorgung und bessere Perspektiven.
Entgeltliche
24.–25. September 2026 | Bonn
Weitere Informationen finden Sie auf der Website der Deutschen Gesellschaft für Humangenetik unter: www.g ev.de

SCARLETRED setzt neue Maßstäbe in der Dermatologie, dank Smartphone-App und künstlicher Intelligenz. Mit Scarletred®Vision ermöglicht das österreichische MedTech-Unternehmen ICH-GCP- und datenschutzkonformes Hautmonitoring aus der Ferne – und verbessert die globale Versorgung seltener Erkrankungen wie Epidermolysis bullosa (EB).

Harald Schnidar CEO und Gründer, SCARLETRED
Eine objektive Dokumentation und Beurteilung von EB-Läsionen ist entscheidend, um den Krankheitsverlauf präzise zu verfolgen und fundierte Therapieentscheidungen zu treffen. Doch für viele Patient:innen, vor allem Kinder, sind häufige Klinikbesuche zeitaufwendig, schmerzhaft und belastend. Scarletred®Vision, ein Softwaresystem entwickelt auf Basis von über zehn Jahren klinischer Forschung und dem KI-Agenten ARORA®, bietet eine sichere, mobile und skalierbare Lösung, die direkt auf Smartphones und somit barrierefrei für alle in grei arer Nähe ist. Im Zentrum steht Scarletred®Vision – eine zertifizierte Software als Medizinprodukt (SaMD), bestehend aus Smartphone-App, Kalibrierungssticker und sicherer Webplattform. Anders als hardwaregebundene Systeme oder nicht zertifizierte Anwendungen ist die Lösung vollständig mobil, zu Hause wie in der Klinik einsetzbar und erfüllt höchste internationale Standards. Führende BioPharma-Unternehmen, akademische Einrichtungen und Ärzt:innen nutzen sie bereits weltweit – in über 100 klinischen Studien und der Marktüberwachung zugelassener Medikamente.
Seltene Erkrankung EB im Fokus Diese Innovation ist besonders wichtig für EB – eine seltene genetische Erkrankung, die etwa eine von 50.000 Geburten betrifft, mit weltweit rund 500.000 Patient:innen. Die sogenannten „Schmetterlingskinder“ leben mit extrem empfindlicher Haut, chronischen Wunden, Infektionsrisiken und oft schweren Komplikationen. Trotz Fortschritten durch Forschung und Patientenorganisationen bleibt
die Versorgung aufwendig und inkonsistent, während Ärzt:innen und Familien oft verlässliche telemedizinische Werkzeuge fehlen.
Automatisiertes EB-Monitoring und -Assessment mit ARORA® Hier setzt SCARLETRED an: Scarletred®Vision mit dem KIAssistenten ARORA® liefert hochqualitative Bilddaten, segmentiert Läsionen automatisch, erkennt Wunden, klassifiziert Gewebearten und verfolgt Blasenbildungen im Verlauf. Ein einziges kalibriertes Smartphone-Foto wird zu einem zuverlässigen klinischen Datensatz – subjektive Beschreibungen gehören der Vergangenheit an. Mit seiner mobilen 2D- und 3D-Imaging-Technologie kann SCARLETRED Blasen schon im Entstehen erkennen. Die Software lokalisiert Blasen nicht nur, sondern beurteilt auch deren Stadium und misst Fläche, Tiefe und Volumen. Scarletred®Vision ermöglicht eine frühere Behandlung sowie eine verbesserte Überwachung von Wundverlauf und Heilung.
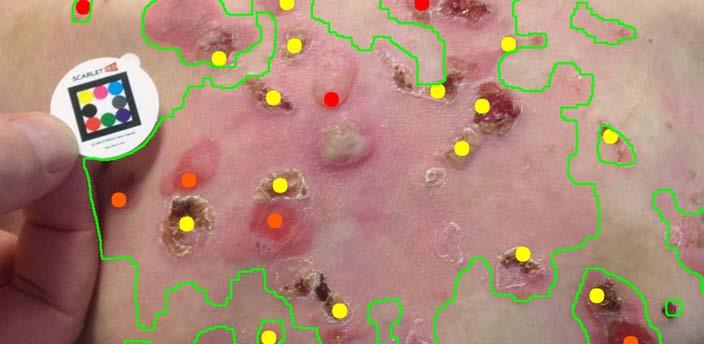
Automatisierte Analyse von EB-Läsionen inklusive Blasenerkennung, Flächensegmentierung, -klassifizierung und 3D-Messung.
Kooperation und praxisnahe Umsetzung
Um die App optimal auf aktuelle Bedürfnisse bei EB anzupassen, arbeitet SCARLETRED bereits langjährig mit führenden Forschungseinrichtungen wie dem Universitätsklinikum Salzburg
(SALK) und dem NIH sowie auch mit Patientenorganisationen zusammen. Studien und Feedbacks von Patient:innen belegen die hohe Nutzerfreundlichkeit und das Vertrauen in die Datensicherheit – eine wesentliche Grundlage für KI-gestützte Entscheidungstools in klinischen Studien und in der Telemedizin. Neben der Unterstützung von BioPharmaUnternehmen und Forscher:innen in der Entwicklung neuer EB-Arzneimittel sieht Harald Schnidar seine Mission vor allem darin: „Bei seltenen Erkrankungen wie EB, bei denen herkömmliche Werkzeuge an ihre Grenzen stoßen, liegt zertifizierte medizinische KI nicht fern in der Zukunft. Gemeinsam mit unseren Partnern und den Patient:innen bringen wir medizinische Innovation dorthin, wo Patient:innen den größten Teil ihres Lebens verbringen – nicht im Krankenhaus, sondern im familiären Umfeld, zu Hause.“
Hier geht’s zur Scarletred®Vision-App: scarletred.com/ products/vision






























Herta S.















































86 Jahre | Zeitzeugin












„Es waren immer Tage des Glücks, wenn ein kam.“
















































Spenden Sie auf care.at










































































Mit freundlicher Unterstützung von Deciphera (Austria) GmbH

Tenosynoviale Riesenzelltumoren (TGCT), auch bekannt als PVNS (pigmentierte villonoduläre Synovitis), sind selten, meist gutartig und entwickeln sich an Gelenken. Wie sie diagnostiziert und behandelt werden, berichtet Priv.-Doz.in Dr.in med. univ. Joanna Szkandera, Onkologin an der Medizinischen Universität Graz.

Priv.-Doz. in Dr. in
med. univ. Joanna Szkandera Programmdirektorin für Knochen- und Weichteilsarkome, Klinische Abteilung für Onkologie der Medizinischen Universität Graz
Was sind TGCT?
Das sind meist gutartige Tumoren, die sowohl in Gelenken als auch außerhalb derselben mit Befall von Sehnenscheiden und Schleimbeuteln wachsen. Warum sie entstehen, ist noch nicht vollständig erforscht. Eine Schlüsselrolle spielt eine Überexpression des Colonystimulating Factor 1 (CSF1).
Welche TGCT gibt es?
Wir unterscheiden lokalisierte (90 Prozent der Fälle) und diffuse TGCT. Lokalisierte TGCT finden sich oft in/an Knie- und kleineren Gelenken wie Hand- und Fingergelenken. Die knotigen Tumoren sind gut abgrenzbar und damit gut operabel. Das Risiko, dass sie nach einer gelungenen OP wiederkehren, ist niedrig. Diffuse TGCT dagegen entwickeln sich häufig in/an Knie-, Hüft- und Sprunggelenken. Sie sind oft aggressiver und zerstörerischer. Wegen ihrer flächig-diffusen Beschaffenheit sind sie nur schwer bis gar nicht zu operieren. Das Risiko, dass durch eine OP die Funktion des betroffenen Gelenks beeinträchtigt wird, ist hoch. Ebenso das Risiko, dass ein diffuser TGCT wiederkehrt.
Wie zeigen sich TGCT? TGCT machen anfangs keine Beschwerden. Wenn diese im weiteren Verlauf auftreten, sind sie unspezifisch: Schwellungen, Schmerzen, Gelenkstei eit und damit Einschränkungen der Beweglichkeit. Das kann die Betroffenen in ihrem Alltag stark beeinträchtigen.
Wen kann die seltene Erkrankung treffen?
Million Menschen treten laut einer europäischen Studie auf. Typischerweise sind Betroffene zwischen 30 und 50 Jahre alt. Frauen erleiden etwas häufiger lokalisierte TGCT, bei diffusen ist das Verhältnis zwischen den Geschlechtern eher ausgeglichen.
Wie diagnostizieren Sie TGCT, und worin besteht dabei die Herausforderung?
Eine zweifelsfreie Diagnose wird oft erschwert beziehungsweise verzögert durch die Seltenheit, die unspezifische Symptomatik und die Verwechslungsgefahr mit Arthrose, Gelenkinnenhautentzündung (Synovitis), Sehnenscheidenentzündung und Meniskuserkrankungen.
Besteht jedoch ein klinischer Verdacht, lässt sich die Diagnose mittels bildgebender Verfahren wie einer Magnetresonanztomografie (MRT) mit Kontrastmittel und histologischen Befunds (Gewebeprobe, Biopsie) stellen. Wobei es immer auf die Erfahrung der Radiolog:innen und Patholog:innen ankommt. Denn per Röntgen oder Ultraschall sind insbesondere diffuse TGCT nur schwer bis gar nicht auszumachen.
Wie entscheidend ist eine möglichst frühe Diagnose?
Die Tumoren wachsen beständig weiter, sodass die befallenen Gelenke immer größeren Schaden nehmen können. Je früher diagnostiziert und gegebenenfalls behandelt wird, desto besser ist die Prognose.
Wie behandeln Sie TGCT – und mit welchem Erfolg?
gewöhnlich erfolgreich operiert. Bei diffusen ist das nicht immer möglich. Bei einigen Patient:innen mit TGCT reicht es aus, die Erkrankung zunächst nur regelmäßig mit MRT-Kontrollen zu beobachten, vor allem wenn wenig oder keine Beschwerden bestehen. Wenn Schmerzen, Schwellungen oder Bewegungseinschränkungen stärker werden, wird in der Regel eine Operation empfohlen, bei der das krankhafte Gewebe im Gelenk möglichst vollständig entfernt wird (Schlüsselloch-OP oder offene Operation). Bestrahlung und Vereisungsbehandlungen werden heute normalerweise nicht mehr eingesetzt, weil der Nutzen unklar ist und die möglichen Nebenwirkungen relevant sind. Wenn eine Operation nicht möglich ist oder mit zu großen Risiken verbunden wäre, können in spezialisierten Zentren gegebenenfalls auch Medikamente eingesetzt werden, die gezielt in den Wachstumsmechanismus des Tumors eingreifen und Beschwerden sowie Tumorgröße verringern können.
Was können Menschen tun, die möglicherweise von TGCT betroffen sind?
Weitere Informationen fi nden Sie unter: livingwithtgct.eu/de DCPH-P03081 I Erstellungsdatum: Februar
Nur geschätzt 5-45 Fälle je eine
Lokalisierte TGCT werden für
Gehen Sie mit Gelenkbeschwerden zu Ärzt:innen, insbesondere wenn Sie sich diese nicht erklären können und sie vier bis sechs Wochen anhalten. Wenn Röntgen und Ultraschall nichts ergeben, fragen Sie gezielt nach einem MRT. Lassen Sie sich gegebenenfalls in ein spezialisiertes Zentrum überweisen. Dort sind multidisziplinäre Teams, die abklären können, ob Sie von einem TGCT betroffen sind, und die Therapieoptionen mit Ihnen besprechen.
Das österreichische Gesundheitswesen steht derzeit unter großem Druck. Budgets werden gekürzt, Medikamente und Therapien immer öfter nicht genehmigt, und vielerorts herrscht ein massiver Sparzwang. Was für viele Patientinnen und Patienten schon jetzt eine Herausforderung ist, trifft Menschen mit seltenen Erkrankungen besonders hart.
In Österreich leben rund 450.000 Menschen mit einer seltenen Erkrankung. Ihre Krankheiten sind oft komplex, schwer zu diagnostizieren und erfordern spezialisierte Behandlungen, die nicht immer leicht zugänglich sind. Viele Betroffene kämpfen jahrelang um eine Diagnose und verlieren wertvolle Zeit. Noch schwieriger wird es, wenn dann dringend benötigte Therapien nicht bewilligt werden oder neue Medikamente mangels Budgets gar nicht erst den Weg zu den Patientinnen und Patienten finden. Zusätzlich hören wir derzeit von einer Häufung an Fällen, in denen Betroffene für bereits in der Vergangenheit mehrfach bewilligte Therapien nun Ablehnungen erhalten.
Pro Rare Austria fordert daher, dass seltene Erkrankungen endlich den Stellenwert bekommen, den sie verdienen. Unsere Hauptanliegen richten sich an Politik, Gesundheitskassen und Entscheidungsträger:innen – mit dem Ziel, eine faire, transparente und nachhaltige Versorgung sicherzustellen.
Wir möchten Politik und Gesellschaft daran erinnern, dass Solidarität im Gesundheitswesen nicht bei der Mehrheit au ören darf. Auch seltene Erkrankungen sind Teil unseres gemeinsamen Gesundheitssystems – und die, die davon betroffen sind, verdienen dieselbe Chance auf ein würdevolles, möglichst lebenswertes Leben
wie alle anderen. Mit klaren politischen Entscheidungen zur Finanzierung, vorausschauender Planung und strukturellen
Reformen ist im Gesundheitssystem langfristig eine Entlastung möglich.
KERNFORDERUNGEN VON PRO RARE AUSTRIA:
1. Nationaler Aktionsplan für seltene Erkrankungen: Der bestehende Plan ist zu wenig wirksam und muss dringend erneuert und mit klaren Zuständigkeiten sowie ausreichendem Budget ausgestattet werden. Nur so kann sichergestellt werden, dass Betroffene in allen Bundesländern gleich gute Chancen auf Diagnose und Therapie haben.
2. Zugang zu Therapien und Medikamenten sichern: Immer häufiger scheitert der Zugang zu innovativen Medikamenten an komplizierten Bewilligungsverfahren oder an der Ablehnung durch Kostenträger. Pro Rare Austria fordert ein transparentes, rasches und patient:innenorientiertes Verfahren, das medizinische Notwendigkeit über Kostenstellen stellt.
3. Finanzielle Absicherung und strukturelle Unterstützung: Forschung, Diagnosezentren und spezialisierte Versorgungsstellen brauchen stabile finanzielle Grundlagen. Der derzeitige Sparkurs gefährdet die Versorgungssicherheit – nicht nur bei seltenen Erkrankungen, aber dort mit besonders dramatischen Folgen.
4. Datensammlung und Forschung fördern: Ohne verlässliche Daten kann keine gute Versorgungsplanung stattfinden. Österreich braucht ein nationales Register, das Forschung, Diagnose und Therapieentwicklung erleichtert und damit langfristig auch Kosten senkt.
5. Bessere Information und Vernetzung: Viele Betroffene irren im Gesundheitssystem umher, ohne eine Ansprechstelle zu finden. Pro Rare Austria fordert den Ausbau von Informationszentren und Anlaufstellen, um Patient:innen, Angehörige und medizinisches Personal zu unterstützen.

Unabhängiger redaktioneller Beitrag
Das erklärt Assoc. Prof.in Priv.-Doz.in Dr.in med. Constanze Jonak, Hautärztin an der Universitätsklinik für Dermatologie der Medizinischen Universität Wien.
Womit bekommen es CTCL-Betroffene zu tun?
CTCL, primär kutane T-ZellLymphome, sind seltene Krebserkrankungen, die von bestimmten weißen Blutkörperchen – den T-Lymphozyten – ausgehen und in erster Linie die Haut betreffen. Die häufigste Variante ist die Mycosis fungoides (MF), das Sézary-Syndrom (SS) hingegen eine sehr seltene mit Haut-, Lymphknoten- und Blutbeteiligung. CTCL-Erkrankte leiden an einer seltenen und sichtbaren Hautkrebserkrankung, die häufig mit einem quälenden Juckreiz einhergeht und die Lebensqualität beträchtlich einschränken kann.
Wie wird die Diagnose gestellt –und was erschwert sie?
Die Diagnose wird klinisch anhand charakteristischer Hautveränderungen und histopathologisch mit mikroskopischer Begutachtung einer Hautprobe gestellt. Auch das Blut – und bei Auff älligkeiten Lymphknoten oder Organe – wird mittels Bildgebung und/oder Gewebeanalysen untersucht. Bei einer MF zeigen sich anfangs meist rote Flecken an der Haut, die anderen Hautkrankheiten wie Schuppenflechte oder Neurodermitis ähneln können, was die Diagnose verzögern und auch zu Fehldiagnosen führen kann. Im weiteren Verlauf kann es zu erhabenen Hautveränderungen und auch Knoten/ Geschwülsten (Tumoren) kommen. Das SS hingegen zeigt sich klinisch mit einer großflächigen Rötung der Haut – einer Erythrodermie, von der man ab 80 Prozent betroffener Körperoberfläche spricht.
Wie behandelt man CTCL?
Aktuell sind CTCL nicht heilbar (abgesehen von Einzelfällen durch Fremdstammzellspenden), aber die Prognose für die meisten Patient:innen, vor allem in frühen Stadien der Erkrankung, ist gut. Die Behandlung soll Symptome lindern, die Lebensqualität verbessern und die Tumorlast reduzieren.

Assoc. Prof. in Priv.-Doz. in Dr. in med. Constanze Jonak Fachärztin für Haut- und Geschlechtskrankheiten, Oberärztin an der Universitätsklinik für Dermatologie in Wien, Leiterin der Ambulanz für kutane Lymphome und der Immundermatologischen Ambulanz
Je nach Krankheitsstadium – ob früh oder fortgeschritten – werden individuell hautgerichtete Therapien, wie Cremes/Salben, Licht- oder Strahlentherapie, und/ oder Systemtherapien, das heißt immunmodulierende, zielgerichtete oder Chemotherapien, eingesetzt.
Was ist der Unterschied zwischen früher und fortgeschrittener Erkrankung?
Frühe Krankheitsstadien sind auf die Haut beschränkt und zeigen sich in roten, mitunter schuppigen Flecken, die im Hautniveau liegen oder leicht erhaben sind. Bei fortgeschrittener Erkrankung treten zudem Hauttumoren oder Erythrodermie auf und es können Lymphknoten, innere Organe und das Blut betroffen sein.
CTCL-Management 2026 versus 2016: Was hat sich verbessert? Wir verfügen heute über eine Antikörper-Toxin-Verbindung (gegen das Oberflächenmerkmal
CD30 gerichtet und mit einem Spindelgift gekoppelt) für CTCL, die dieses Oberflächenmolekül tragen. Außerdem steht ein gegen CCR4, die Andockstelle auf T-Zellen, gerichteter Antikörper für die Behandlung von MF/SS nach jeweils einer vorangegangenen Systemtherapie zur Verfügung sowie eine örtliche Chemotherapie in einer Gelformulierung. Diese Therapien haben die CTCL-Behandlung revolutioniert, dennoch sind Dauerremissionen, also langandauernde Krankheitsfreiheit, selten.
An wen können sich CTCL-Patient:innen wenden, um ihre Erkrankung besser zu bewältigen?
Die Diagnose, Behandlung und auch Au lärung über die Erkrankung sollte von Expert:innen an spezialisierten Zentren durchgeführt werden. Die Selbsthilfegruppe für Hautlymphome bietet Betroffenen und Angehörigen Erfahrungsaustausch und Unterstützung.
Lesen Sie mehr unter hautlymphome.org
E-Mail: selbsthilfe. hautlymphome@ gmx.at
Der Übergang von der Kinder- in die Erwachsenenmedizin als partizipativer, interdisziplinärer und vernetzter Prozess
Die Österreichische Liga für Kinder- und Jugendgesundheit beschäftigt sich als Netzwerkorganisation mit über 120 Mitgliedsorganisationen seit 2017 mit dem Thema Transition, veranstaltet dazu regelmäßige Dialogformate und betreibt die Videoplattform Transition: www.kinderjugendgesundheit.at/projekte/ videoplattform-transition
DAngelika Heumader-Rainer
Geschäftsführerin der Österreichischen Liga für Kinder- und Jugendgesundheit
FOTO: ZVG
ank erheblicher medizinischer Fortschritte erreichen heute immer mehr Kinder mit chronischen und seltenen Erkrankungen das Erwachsenenalter – auch bei Diagnosen, für die noch vor wenigen Jahrzehnten kaum Therapien zur Verfügung standen. Damit rückt der altersbedingte Wechsel von der kinderzentrierten in die erwachsenenorientierte Versorgung („Transition“) zunehmend in den Fokus. Dieser Übergang ist jedoch mit vielfältigen Herausforderungen verbunden und bedarf gemeinsamer Anstrengungen und vernetzter Konzepte, um zu gelingen.
Erwachsenwerden als mehrfach kritische Phase Junge Menschen mit seltenen oder chronischen Erkrankungen werden meist in hoch spezialisierten pädiatrischen Strukturen betreut. Über Jahre hinweg werden sie von interdisziplinären Teams begleitet, mit festen Ansprechpartner:innen und umfassender Expertise. Mit der Volljährigkeit fällt für sie die ohnehin komplexe Lebensphase des Erwachsenwerdens mit einer tiefgreifenden medizinischen Umstellung zusammen: Bildungsund Berufsentscheidungen, Ablösung vom Elternhaus und Identitätsentwicklung treffen auf den Abschied von vertrauten Behandlungsteams.
Das kalendarische Alter allein kann dabei kein objektiver Maßstab für den Übertritt sein. Der geeignete Zeitpunkt hängt von Erkrankung, Entwicklungsstand und individueller Lebenssituation ab. Fachleute unterscheiden zudem zwischen dem „Transfer“ als – im Idealfall individuellem – Stichtag des Wechsels und der „Transition“ als langfristigem Prozess, der idealerweise bereits
im frühen Jugendalter beginnt und über Jahre begleitet wird.
Partizipation und Empowerment
Betroffene betonen die Bedeutung einer frühzeitigen, offenen Thematisierung ihrer Erkrankung und echter Mitentscheidung. Zentral ist dafür die schrittweise Befähigung junger Patient:innen, ihre Erkrankung zu verstehen und Verantwortung zu übernehmen. Dazu gehören Wissen über Diagnose und Therapie, eigenständige Terminorganisation sowie die Fähigkeit, mit unterschiedlichen Versorgungssystemen zu kommunizieren. Für einzelne Indikationen existieren hierzu bereits altersgerechte Informationsmaterialien, Bücher, Videos oder strukturierte Gesprächsleitfäden, die diesen Prozess unterstützen.
Ebenso wichtig ist die Rolle der Eltern: Über viele Jahre hinweg agieren sie als Co-Manager:innen der Erkrankung. Der Übergang zu mehr Autonomie sollte daher früh von den Erziehungsberechtigten reflektiert und bewusst gestaltet werden – mit dem Ziel größtmöglicher Selbstständigkeit und Selbstbestimmung der jungen Erwachsenen.
Kontinuität sichern, Wissen erhalten und/oder transferieren In der Erwachsenenmedizin fehlt häufig die Spezialisierung auf seltene oder ursprünglich primär pädiatrische Krankheitsbilder. Gerade bei komplexen Diagnosen droht dadurch ein Wissens- und Betreuungsvakuum. Um Versorgungskontinuität sicherzustellen, braucht es daher klar definierte Verantwortlichkeiten, strukturierte Übergabeprozesse sowie die Verankerung von Transition in
Ausbildung und klinischer Praxis. Hilfreich können hier digital gestützte Koordinationstools, evidenzbasierte Leitlinien und international abgestimmte Standards sein. Auch interdisziplinäre Teams mit eigenen TransitionKoordinator:innen gewinnen an Bedeutung. Beispielhaft wird am Wiener Zentrum für seltene und unbekannte Erkrankungen (CCRUD) das krankheitsübergreifende Programm „Ready4Transition“ umgesetzt. Es umfasst checklistenbasierte Begleitung, psychosoziale Gesprächsleitfäden und Instrumente zur Förderung der Selbstkompetenz.
Navigation und Vernetzung
Wenn geeignete Versorgungspendants in der Erwachsenenmedizin existieren, bleibt die Herausforderung, diese ausfindig zu machen. Zeitliche, geografische oder finanzielle Hürden können zusätzlich den Zugang erschweren. Hier leisten Selbsthilfe- und Patient:innenorganisationen wertvolle Unterstützung, indem sie – oftmals spezifische und damit zielgenaue – Informationen bündeln, Kontakte vermitteln und Orientierung bieten.
Auch digitale Vernetzungsmöglichkeiten eröffnen niederschwellige Zugänge und großes Informationspotenzial. Gleichzeitig betonen Betroffene und Fachkräfte die Unersetzbarkeit persönlicher Austauschformate. Regelmäßige Vernetzungstreffen fördern Wissenstransfer, stärken Kooperationen und ermöglichen eine gemeinsame Weiterentwicklung von Strukturen. Vor diesem Hintergrund lädt die Kinderliga im Dezember diesen Jahres zum Symposium Transition ein.
Vertreter:innen aus Medizin, Pflege, Psychologie, Sozialarbeit, Gesundheitspolitik, Selbsthilfe und Patient:innenvertretung diskutieren, wie Transition künftig noch stärker vernetzt und patient:innenorientiert und nachhaltig gestaltet werden kann. Ziel ist es, Erfahrungen zu bündeln, Modelle weiterzuentwickeln und konkrete Strategien für eine strukturierte, interdisziplinäre und partizipative Transition zu erarbeiten.
Gemeinsam. Partizipativ. Interdisziplinär.
Save the Date 4. Dezember 2026 Wien
1.
Zellmodelle und Tierversuche
Bevor neue Wirkstoffe in klinischen Studien, und damit an Menschen, geprüft werden, liefern präklinische Studien erste Hinweise über Wirkung, Verträglichkeit und Dosierung. Dies passiert im Labor an Zellmodellen oder über Tierversuche. Nur wenn keine gefährlichen Nebenwirkungen auftreten, werden Wirkstoffe anschließend in klinischen Studien getestet.
Allgemeine Verträglichkeit
Unter strenger ärztlicher Überwachung wird in der Phase I an gesunden freiwillig teilnehmenden Menschen ein neuer Wirkstoff hinsichtlich der allgemeinen Verträglichkeit geprüft. Dabei werden unter anderem Informationen zu Anwendung, Aufnahme und Ausscheidung gesammelt.
4.
Breite Erprobung des Wirkstoffes
Die Phase III steht für die große und breite Prüfung eines neuen Wirkstoffes. In dieser Phase, die häufig in mehreren Ländern zugleich stattfindet, wird der neue Wirkstoff an Hunderten, teilweise sogar Tausenden Patient:innen getestet. Wird die Phase III erfolgreich abgeschlossen, kann die Zulassung des neuen Medikaments bei den jeweiligen Arzneimittelbehörden eingereicht werden.
3.
Verträglichkeit bei Patient:innen
In der Phase II stehen die Dosierung und die Verträglichkeit bei Patient:innen im Fokus. Damit erhält die klinische Forschung Erkenntnisse über die Wirkung der neuen Substanz in kranken Organismen. Grundsätzlich wird der Wirkstoff mit einer Kontrollsubstanz, einem „Placebo“, verglichen, wobei weder Patient:innen noch Ärztinnen oder Ärzte wissen, wer den neuen Wirkstoff oder das Placebo erhält.
5.
Weitere klinische Überprüfungen
Nach der Zulassung können in Phase-IV-Studien weiterhin Daten zu Wirksamkeit und Verträglichkeit gesammelt werden, mit dem Ziel, dringend benötigte Medikamente für schwere, fortschreitende Erkrankungen Patient:innen schneller zugänglich zu machen.
Der Weg von einem Wirkstoff bis hin zu einem zugelassenen Arzneimittel ist aufwendig, langwierig und mit hohen Kosten verbunden. Von etwa 5.000 bis 10.000 Substanzen, die in der Arzneimittelforschung untersucht werden, gelten nach rund fünf Jahren im Durchschnitt nur noch etwa neun als vielversprechend. Nach der Präklinik sowie umfangreichen klinischen Prüfungen bleibt meist nur eine einzige Substanz übrig, die schließlich als neue medikamentöse Therapie zugelassen wird. Insgesamt dauert die Entwicklung eines Arzneimittels im Schnitt etwa 13 Jahre.
Die Entwicklung eines neuen, innovativen Medikaments kostet durchschnittlich bis zu 2,6 Milliarden US-Dollar. Das liegt nicht nur an der aufwendigen Forschung, sondern auch daran, dass viele Projekte scheitern. Zusätzlich entstehen hohe Kosten durch strenge Sicherheits- und Dokumentationsvorgaben, steigende medizinische Standards und große klinische Studien mit vielen Teilnehmenden.
Forschung
Präklinik
UNSER AUFTR AG FÜ R INN OVATI V E LÖSUNG E N

TAG DER Am 28. Februar ist
Seltenen Erkrankungen
TAKEDA UNTERSTÜTZT MENSCHEN MIT SELTENEN UND KOMPLEXEN ERKRANKUNGEN
Fünf Prozent der Weltbevölkerung leiden an Seltenen Erkrankungen.1 In Österreich sind 450.000
2 Takeda unterstützt die Patient*innen von der Diagnose bis zur bestmöglichen Versorgung mit Therapien. Seit 70 Jahren entwickeln und produzieren wir in Österreich eine Vielzahl
1 Global Genes RARE Diseases: Facts and Statistics. Verfügbar unter: https://globalgenes.org/rare-disease-facts/ Letzter Zugri : Februar 2026
2 Dachverband Pro Rare Austria Verfügbar unter: https://www prorare-austria org/pro-rare-austria/wer-wir-sind Letzter Zugriff: Februar 2026
ERFAHREN SIE MEHR über den Kranich und das Engagement von Takeda für Menschen mit Seltenen Erkrankungen
www.takeda.at