Associate Professor of Ophthalmology and Neurology
Director of Neuro-Ophthalmology Service
Department of Ophthalmology and Visual Sciences
University of Iowa Iowa City, IA
Robert L. Tomsak, MD, PhD
Professor of Ophthalmology and Neurology
Kresge Eye Institute
Wayne State University Detroit, MI
1
Oxford University Press is a department of the University of Oxford. It furthers the University’s objective of excellence in research, scholarship, and education by publishing worldwide. Oxford is a registered trade mark of Oxford University Press in the UK and certain other countries.
Published in the United States of America by Oxford University Press
198 Madison Avenue, New York, NY 10016, United States of America.
All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, without the prior permission in writing of Oxford University Press, or as expressly permitted by law, by license, or under terms agreed with the appropriate reproduction rights organization. Inquiries concerning reproduction outside the scope of the above should be sent to the Rights Department, Oxford University Press, at the address above.
You must not circulate this work in any other form and you must impose this same condition on any acquirer.
Library of Congress Cataloging-in-Publication Data
Names: Thurtell, Matthew J., author. | Tomsak, Robert L., author.
Title: Neuro-ophthalmology / by Matthew J. Thurtell, Robert L. Tomsak.
Description: Second edition. | New York, NY : Oxford University Press, [2019] | Includes bibliographical references and index.
Identifiers: LCCN 2018054874 | ISBN 9780190603953 (pbk.)
Subjects: | MESH: Eye Diseases—diagnosis | Nervous System Diseases—complications | Cranial Nerve Diseases | Eye Diseases—therapy | Diagnosis, Differential | Case Reports Classification: LCC RE75 | NLM WW 460 | DDC 617.7075—dc23 LC record available at https://lccn.loc.gov/2018054874
This material is not intended to be, and should not be considered, a substitute for medical or other professional advice. Treatment for the conditions described in this material is highly dependent on the individual circumstances. And, while this material is designed to offer accurate information with respect to the subject matter covered and to be current as of the time it was written, research and knowledge about medical and health issues is constantly evolving and dose schedules for medications are being revised continually, with new side effects recognized and accounted for regularly. Readers must therefore always check the product information and clinical procedures with the most up-to-date published product information and data sheets provided by the manufacturers and the most recent codes of conduct and safety regulation. The publisher and the authors make no representations or warranties to readers, express or implied, as to the accuracy or completeness of this material. Without limiting the foregoing, the publisher and the authors make no representations or warranties as to the accuracy or efficacy of the drug dosages mentioned in the material. The authors and the publisher do not accept, and expressly disclaim, any responsibility for any liability, loss or risk that may be claimed or incurred as a consequence of the use and/or application of any of the contents of this material.
9 8 7 6 5 4 3 2 1
Printed by WebCom, Inc., Canada
Robert B. Daroff, MD—our mentor, colleague, and friend—in recognition of his contributions to the field of neuro-ophthalmology.
14 Visual Auras, Hallucinations, and Illusions 77 15 Transient Vision Loss 83 16 Unexplained Vision Loss 89 17 Nonorganic Vision Loss 93 SECTION II EFFERENT DISORDERS 18 Third Nerve Palsy 99 19 Fourth Nerve Palsy 105
20 Sixth Nerve Palsy 111
21 Intermittent Diplopia 115
22 Ocular Myasthenia 121
23 Infranuclear Ophthalmoplegia 127
24 Internuclear Ophthalmoplegia 133
25 Supranuclear Ophthalmoplegia 137
26 Gaze-evoked Nystagmus 141
27 Downbeat Nystagmus 145
28 Upbeat Nystagmus 151
29 Pendular Nystagmus 155
30 Infantile Nystagmus 161
31 Saccadic Intrusions and Dysmetria 167
SECTION III EYELID DISORDERS
32 eyelid Ptosis 175
33 Benign essential Blepharospasm 181
SECTION IV PUPIL DISORDERS
34 Anisocoria 187
35 Horner Syndrome 193
36 Tonic Pupil 199
SECTION V ORBITAL AND MISCELLANEOUS DISORDERS
37 Thyroid eye Disease 207
38 Syndromes of the Orbital Apex, Superior Orbital Fissure, and Cavernous Sinus 213
39 Carotid-Cavernous Fistula 219
40 Dorsal Midbrain Syndrome 225
Index 231
Preface
Patients with neuro-ophthalmic conditions are commonly encountered in clinical practice, yet many clinicians feel ill prepared or uncomfortable when dealing with them. Those trained in neurology often feel uneasy when evaluating patients who have predominantly visual or ocular complaints, whereas those trained in ophthalmology often feel uncomfortable when evaluating those with predominantly neurologic complaints. Since a neuro-ophthalmologist is not always available for consultation, the clinician might be left wondering, “What do I do now?” when encountering a challenging case.
In this installment of the What Do I Do Now? series, we aim to provide a user-friendly manual that clinicians can reference when dealing with patients who have neuro-ophthalmic problems. The volume is divided into five sections that cover the main subdivisions of neuro-ophthalmic practice: (1) afferent (visual) disorders, (2) efferent (eye movement) disorders, (3) eyelid disorders, (4) pupil disorders, and (5) orbital and miscellaneous disorders. Each chapter includes a practical, case-based discussion on how to approach and manage a patient with a particular disorder. We discuss mainly common disorders, although we also consider some less common yet important conditions, as well as neuro-ophthalmic presentations of systemic and psychiatric disease. We have included a large number of efferent (eye movement) cases because many other handbooks focus on afferent (visual) disorders. We have based our recommendations on current evidence whenever possible. A list of key clinical points appears at the end of each chapter as well as a list of important references. In most chapters, tables and boxes summarize pertinent information. We include figures in most chapters to illustrate abnormal clinical signs or relevant imaging findings.
We designed the volume as a resource for neurologists and ophthalmologists at all levels of training. We hope that it will serve as a useful handbook in caring for patients with neuro-ophthalmic disease.
Acknowledgments
Many of the cases described in this book touch on controversial aspects of neuro-ophthalmology. We acknowledge those experts in the field who agreed to discuss their personal approaches to such cases with us. In particular, we thank Drs. Randy H. Kardon, Michael Wall, Richard C. Allen, and Wallace L. Alward for their helpful advice. We also thank the team at Oxford University Press, Craig Panner and Tiffany Lu, for their help in bringing this volume to completion.
SECTION I
Afferent Disorders
1 Optic Neuritis
You are called to see a 22-year-old woman who has had a subacute onset of vision loss in her right eye over several days with associated pain on eye movements. She has no past medical history and denies other neurologic symptoms. Examination shows visual acuities of 20/100 in the right eye and 20/15 in the left eye. She can identify only the control Ishihara color plate with the right eye, but she correctly identifies all plates with the left eye. Confrontation visual fields show a central scotoma in the right eye. Her pupils are equal and react to light, but there is a right relative afferent pupillary defect. Her funduscopic examination is normal.
What do you do now?
The patient in this scenario has had a subacute onset of painful monocular vision loss and exhibits the cardinal signs of an optic neuropathy: decreased visual acuity, a central visual field defect, dyschromatopsia, and a relative afferent pupillary defect. Her presentation is classic for optic neuritis. The clinical manifestations of optic neuritis vary depending on the portion of the optic nerve that is inflamed. For example, in retrobulbar optic neuritis, the most common subtype of optic neuritis, the retrobulbar portion of the optic nerve is inflamed and there is minimal, if any, optic disc edema. However, in neuroretinitis, the optic nerve head and peripapillary retina are inflamed, giving rise to optic disc edema and changes in the macula (i.e., fluid extending into the macula initially with development of retinal exudates to form a macular star) (see Case 7).
Causes for optic neuritis also vary depending on the portion of the optic nerve affected. Causes for retrobulbar optic neuritis include demyelinating diseases (e.g., multiple sclerosis [MS] and neuromyelitis optica), autoimmune diseases (e.g., systemic lupus erythematosus), inflammatory diseases (e.g., sarcoidosis), infections (e.g., Lyme disease and syphilis), and vaccinations (e.g., influenza vaccine). In many patients, however, a cause cannot be identified and the optic neuritis is considered idiopathic. Nevertheless, the first step in the evaluation of the patient described in this scenario would be to obtain further history (e.g., inquiring about recent travel, vaccinations, and tick bites). The ophthalmic and neurologic examinations should be completed, because there may be abnormalities that suggest the diagnosis. For example, the presence of granulomatous uveitis on ophthalmic examination might suggest an inflammatory disorder, such as sarcoidosis, whereas the presence of internuclear ophthalmoplegia might suggest MS. In the absence of any other significant history or abnormal examination findings, however, the optic neuritis is likely to be idiopathic.
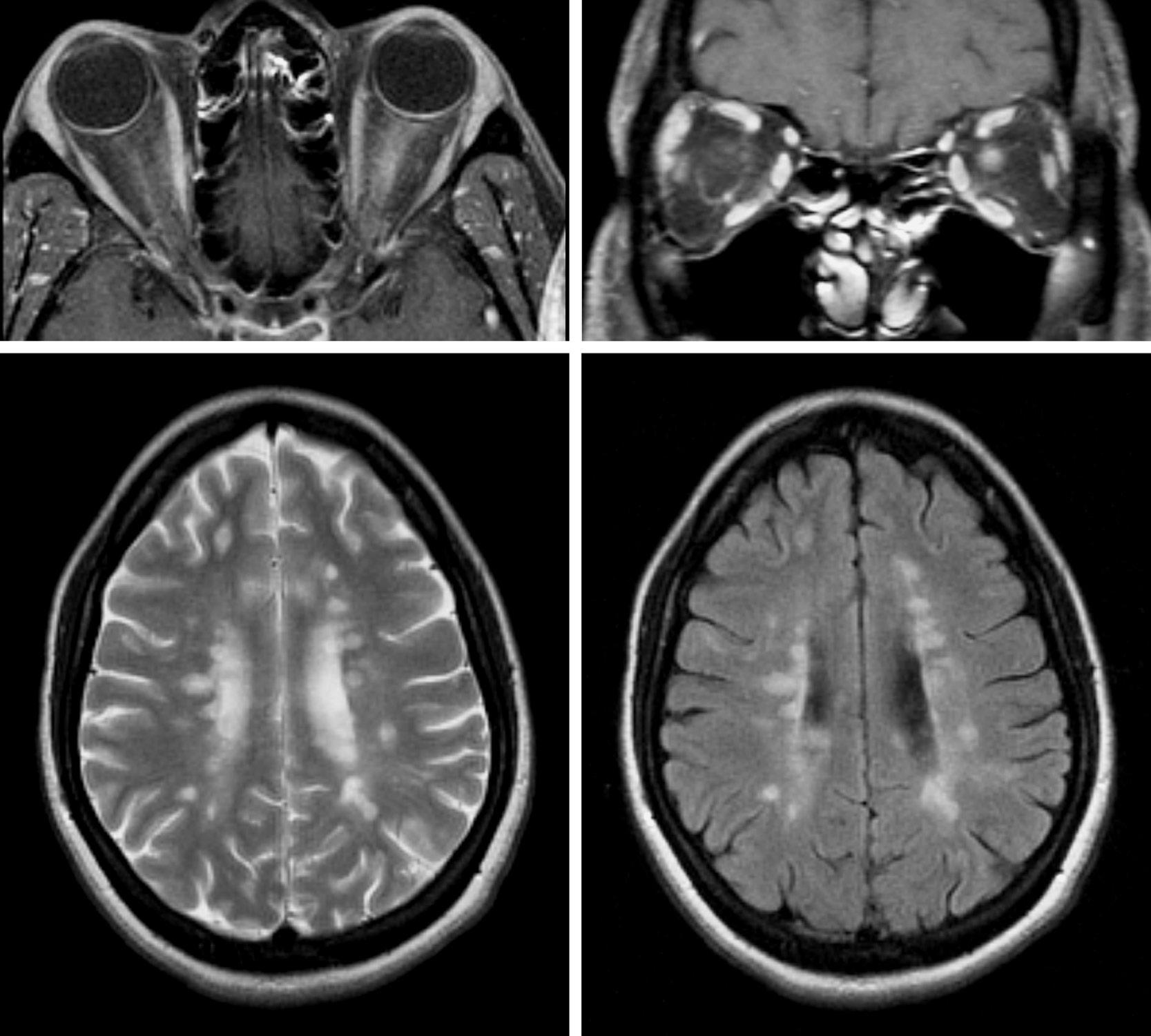
Diagnostic studies should be obtained in patients with idiopathic optic neuritis to confirm the presence of optic nerve enhancement, to evaluate for white matter lesions in the brain, and to exclude other causes for the optic neuropathy (e.g., compressive optic neuropathy; see Case 4). The most important initial diagnostic study is magnetic resonance imaging (MRI) of the orbits and brain. MRI of the orbits typically demonstrates increased signal in the affected optic nerve, with associated contrast enhancement that is best appreciated on fat-suppressed images (Figure 1.1). However, such
changes are nonspecific and cannot be considered diagnostic of idiopathic optic neuritis. MRI of the brain should be obtained to assess for white matter lesions, which are most obvious on the T2-weighted and FLAIR (fluid-attenuated inversion recovery) sequences (see Figure 1.1), because the presence of one or more lesions (especially when they have a periventricular distribution) portends an increased risk of developing MS.
Laboratory investigations can be useful to screen for other causes of optic neuritis, such as neuromyelitis optica (NMO), especially in patients with atypical features to their presentation (Box 1.1). Patients with optic neuritis in the setting of NMO with NMO-IgG (i.e., aquaporin-4) antibodies often present with severe vision loss that can be bilateral and without associated pain, and a poor recovery of vision despite aggressive treatment. Patients
FIGU re 1.1. MRI of the orbits (top row) and brain (bottom row) in a patient with optic neuritis, demonstrating left optic nerve enhancement and multiple ovoid periventricular white matter lesions consistent with MS.
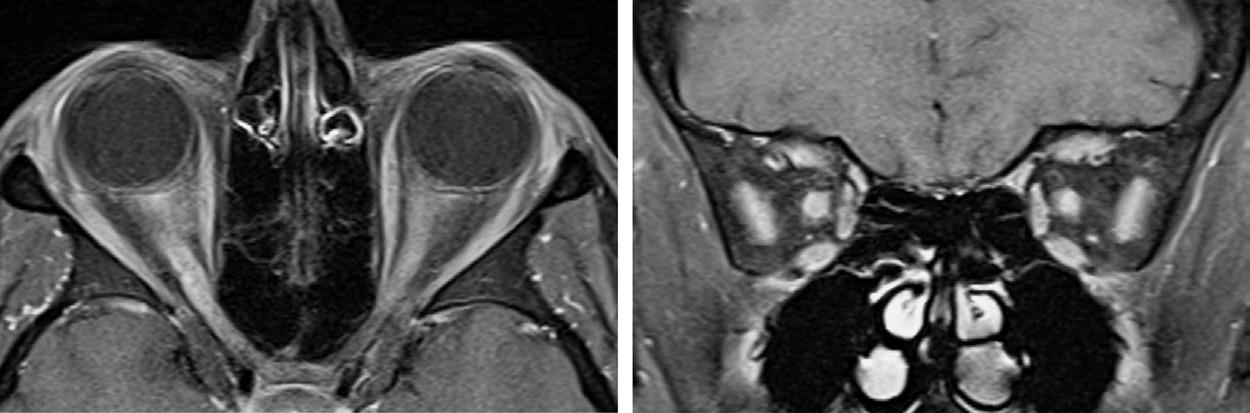
with optic neuritis in the setting of NMO with myelin oligodendrocyte glycoprotein (i.e., MOG-IgG) antibodies can also present with severe vision loss that can be bilateral with associated optic disc edema, but usually have a rapid and dramatic recovery of vision with treatment. With optic neuritis in the setting of NMO, MRI of the orbits can show longitudinally extensive optic nerve enhancement or bilateral optic nerve enhancement (Figure 1.2). Other laboratory investigations, such as antinuclear antibody, antineutrophil cytoplasmic antibodies, angiotensin-converting enzyme level, lysozyme level, syphilis serology, and Lyme serology, are often unrevealing and, thus, unnecessary unless there are atypical features to the presentation (see Box 1.1). Cerebrospinal fluid (CSF) analysis can be useful to evaluate for other causes of optic neuritis in selected patients. However, CSF analysis
BOX 1.1 Atypical Features for Idiopathic Optic Neuritis
Lack of pain
Severe vision loss
Poor recovery of vision
Bilateral optic nerve involvement
Systemic symptoms or signs (e.g., fever or rash)
Intraocular inflammation, hemorrhages, or exudates
Longitudinally extensive optic nerve enhancement on MRI
Optic nerve sheath enhancement on MRI
Rapid improvement following initiation of steroids
Relapsing course following withdrawal of steroids
FIGU re 1.2. MRI of the orbits in a patient with bilateral optic neuritis in the setting of neuromyelitis optica, demonstrating longitudinally extensive bilateral optic nerve enhancement.
is not routinely required to evaluate for oligoclonal bands and other CSF markers of MS, because an increased risk of MS is more reliably predicted by the presence of white matter lesions on MRI.
The recommended treatment of idiopathic optic neuritis is based on the findings of the Optic Neuritis Treatment Trial, which was a randomized controlled trial comparing outcomes in patients who received intravenous and then oral steroids, oral steroids alone, or placebo. The group receiving intravenous and then oral steroids showed a more rapid recovery of vision than the placebo group, although the final visual outcome was similar. This group also showed a lower risk of developing clinically definite MS in the first 2 years following treatment. The group receiving oral steroids alone did not show a faster recovery compared with placebo but did show an increased rate of recurrent optic neuritis attacks compared with the other two groups. Thus, the recommended treatment protocol for idiopathic optic neuritis is intravenous methylprednisone (1 g daily) for 3 days followed by oral prednisone (1 mg/kg daily) for 11 days. The prognosis for visual recovery is excellent, with vision recovering to near normal in most patients over weeks to months, although there can be minor persisting visual deficits (e.g., in contrast and color vision) and clinical signs of optic nerve dysfunction (e.g., a relative afferent pupillary defect and optic disc pallor).
In patients who have one or more white matter lesions on MRI, the risk of developing MS is greater than 65% in the 15 years following an attack of idiopathic optic neuritis, compared with 25% in patients who do not have white matter lesions. Treatment with disease-modifying therapy for MS (e.g., beta-interferon) can reduce the risk of developing MS in patients with idiopathic optic neuritis who have white matter lesions on MRI. Although not all optic neuritis patients with white matter lesions will develop clinically definite MS, initiation of disease-modifying therapy should be carefully considered. K e Y POINTS TO re M e MB er
• The cardinal signs of an optic neuropathy are decreased visual acuity, a central visual field defect, dyschromatopsia, and a relative afferent pupillary defect.
• Optic neuritis is characterized by a subacute onset of painful monocular vision loss with a relative afferent pupillary defect; optic disc edema may or may not be present, depending on the portion of the nerve affected.
• Optic neuritis is often idiopathic but can be caused by demyelinating disease, autoimmune disease, inflammatory disease, infections, and vaccinations.
• MRI of the orbits and brain should be obtained acutely to confirm the presence of optic nerve enhancement, evaluate for white matter lesions in the brain, and exclude other causes of optic neuropathy.
• The 15-year risk of developing MS is over 65% in optic neuritis patients with one or more white matter lesions on MRI versus about 25% in those with no white matter lesions.
• Treatment of optic neuritis with intravenous methylprednisone (1 g daily) for 3 days and then oral prednisone (1 mg/kg daily) for 11 days leads to a faster recovery of vision and decreased risk of developing MS in the 2 years following treatment.
Further Reading
Beck RW, Cleary PA, Anderson MM Jr, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. N Engl J Med. 1992;326:581–588.
Beck RW, Cleary PA, Trobe JD, et al. The effect of corticosteroids for acute optic neuritis on the subsequent development of multiple sclerosis. N Engl J Med. 1993;329:1764–1769.
Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. Myelin oligodendrocyte glycoprotein antibody (MOG-IgG)-positive optic neuritis: clinical characteristics, radiologic clues and outcome. Am J Ophthalmol. 2018;195:8–15.
Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. Arch Neurol. 2008;65:727–732.
Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17:162–173.
2 Arteritic Ischemic
Optic Neuropathy
A 75-year-old white woman presents to your clinic after suddenly developing vision loss in her right eye. She reports having had several brief episodes of transient vision loss in her right eye over the past week, as well as a persistent dull rightsided temporal headache. Examination shows visual acuities of count fingers in the right eye and 20/25 in the left eye. She cannot identify the control Ishihara color plate with the right eye, but she correctly identifies all plates with the left eye. Her pupils are equal and react to light, but there is a right relative afferent pupillary defect. Funduscopic examination shows pallid optic disc edema in the right eye.
What do you do now?
The acute onset of severe monocular vision loss with associated pallid optic disc edema in this older woman with temporal headaches should immediately suggest anterior ischemic optic neuropathy secondary to giant cell arteritis (GCA). Ischemic optic neuropathies occur as a result of hypoperfusion of the optic nerve. Ischemia to the retrobulbar portion of the nerve results in posterior ischemic optic neuropathy; because the anterior portion of the optic nerve is not affected, there is no associated optic disc edema. Ischemia to the anterior portion of the nerve results in anterior ischemic optic neuropathy; because the optic nerve head is affected, there is, by definition, optic disc edema, which can be hyperemic or pallid. Anterior ischemic optic neuropathy occurs more frequently than posterior ischemic optic neuropathy. It can be divided into two types: arteritic anterior ischemic optic neuropathy (AAION), in which the ischemia results from inflammatory narrowing or occlusion of the posterior ciliary arteries by vasculitis (most commonly GCA), and nonarteritic anterior ischemic optic neuropathy (NAION), in which the ischemia occurs because of other factors (see Case 3).
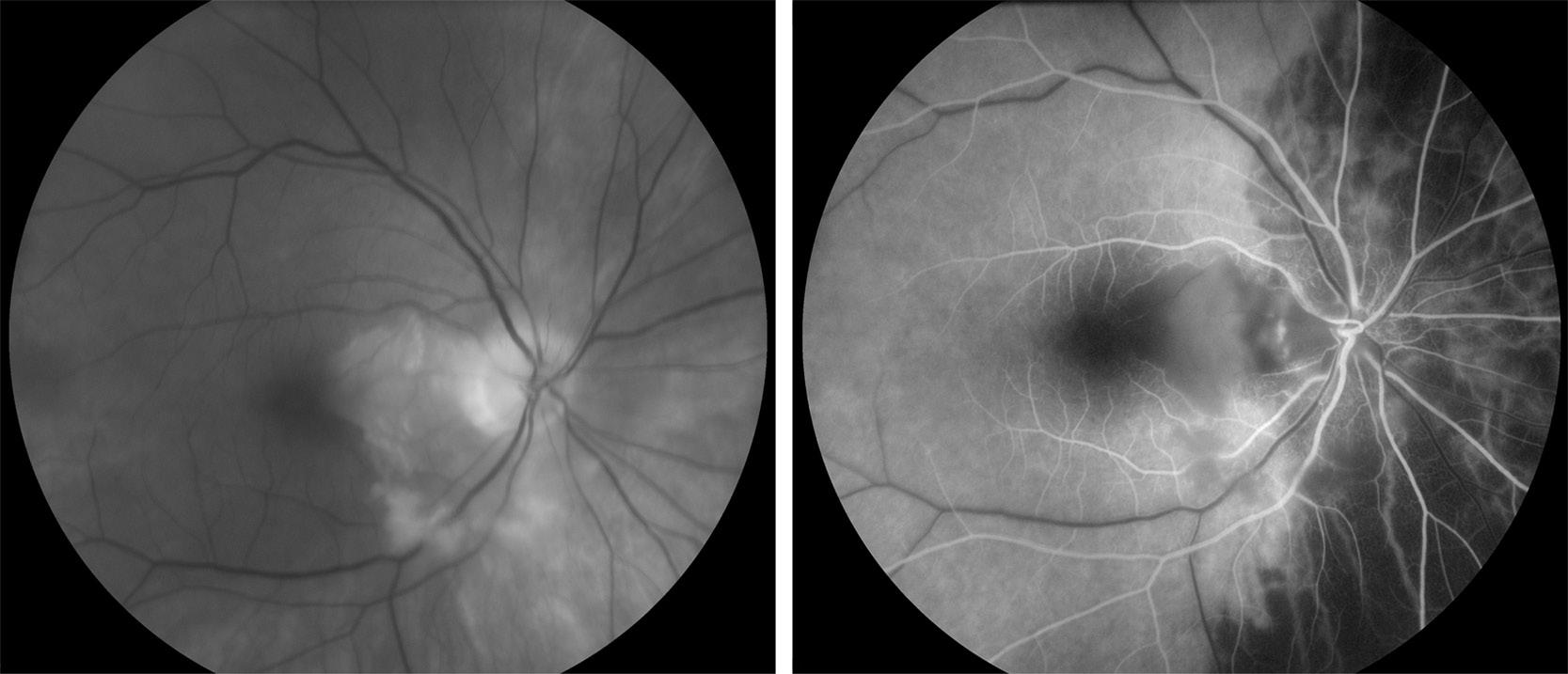
GCA is a granulomatous vasculitis that affects medium- to large-sized arteries, especially the cranial branches of the aortic arch. It occurs most commonly in white women who are aged 65 years or more and does not occur in children or adults who are aged 50 years or less. Up to 50% of patients present with visual symptoms, mostly secondary to AAION. The AAION in GCA is characterized by a rapid onset of severe monocular vision loss with diffuse optic disc edema (Figure 2.1) and a dense relative afferent pupillary defect. The vision loss is often devastating, with the initial visual acuity being count fingers or worse in over 50% of patients. The optic disc edema is typically pallid, but it can be hyperemic. The optic disc edema gradually resolves over several weeks, with the optic disc ultimately becoming pale, atrophic, and cupped. In contrast with optic neuritis and NAION (see Cases 1 and 3), there is rarely any recovery of vision. AAION can sometimes be difficult to distinguish from NAION in the acute setting. The distinction is clinically important, however, because 25% to 50% of patients with AAION will develop AAION in the fellow eye within 2 weeks if left untreated. The presence of pallid optic disc edema is highly suggestive of AAION, whereas hyperemic optic disc edema with retinal nerve fiber layer hemorrhages is more characteristic of NAION (see Case
3). Since GCA causes inflammatory narrowing and occlusion of the posterior ciliary arteries (which supply the choroid of the eye) and the cilioretinal artery (which variably supplies the papillomacular portion of the retina), concurrent choroidal or cilioretinal ischemia is highly suggestive of GCA (see Figure 2.1). Prior to the onset of AAION, a significant proportion of patients will have episodes of transient monocular or binocular vision loss, which are typically precipitated by postural changes. Some patients experience transient diplopia, which is thought to be secondary to ischemia of the extraocular muscles. Many report systemic symptoms, such as temporal headache, jaw claudication, scalp tenderness, malaise, weight loss, and fever, which should immediately suggest GCA. However, their absence does not preclude GCA; over 20% of patients with biopsy-proven GCA do not have systemic symptoms.
For the patient in this scenario, with a dull temporal headache and severe monocular vision loss due to anterior ischemic optic neuropathy, the clinical suspicion for GCA is high. Therefore, urgent investigations and treatment are required. An erythrocyte sedimentation rate and C-reactive protein level should be obtained immediately. Most patients with GCA have elevation of both inflammatory markers, reflecting systemic inflammation, but occasionally only one might be elevated. In patients who do not have elevated inflammatory markers, further investigations should still be obtained and empiric treatment initiated if the clinical suspicion for
FIGU re 2.1. Fundus photograph demonstrating pallid optic disc edema due to AAION, with cilioretinal artery occlusion, in a patient with GCA (left). Fluorescein angiography shows patchy choroidal nonperfusion and cilioretinal artery occlusion (right).
GCA is high. Fluorescein angiography can be useful to demonstrate choroidal or cilioretinal ischemia, thereby helping to differentiate AAION from NAION (see Figure 2.1). However, the diagnosis can be confirmed only by identifying the characteristic histopathologic changes on temporal artery biopsy. The specimen should be at least 2 cm long and serially sectioned, to avoid a false-negative result in the event that there are skip lesions. If the clinical suspicion is low, a negative biopsy is sufficient to exclude the diagnosis. The initiation of treatment should not be delayed while awaiting a temporal artery biopsy or its result; the histopathologic changes persist for at least several weeks after treatment is commenced.
In any patient with suspected AAION, systemic corticosteroids should be administered immediately to reduce the risk of AAION occurring in the fellow eye. Although no prospective randomized study has been performed, many clinicians treat with intravenous methylprednisone (1 g daily) for 1–3 days followed by high-dose oral prednisone (1 mg/kg daily), although some will begin with high-dose oral prednisone alone. High-dose oral prednisone should be continued for at least a month, until systemic symptoms have resolved and the inflammatory markers have normalized. When prednisone is used as monotherapy, its dose should be slowly tapered over 12–18 months (e.g., by 5–10 mg per month), provided that the patient does not have any symptoms to suggest active GCA and his or her inflammatory markers remain within normal limits. Since many patients develop significant complications and side effects from prolonged prednisone therapy (e.g., osteoporosis, steroidinduced diabetes, and weight gain), addition of a steroid-sparing agent (e.g., leflunomide) should be considered. One recent trial found that therapy with tocilizumab, a monoclonal antibody to the interleukin-6 receptor, allowed for a more rapid taper of prednisone with less serious adverse events than therapy with prednisone alone. However, long-term follow-up is needed to determine the durability of remission with tocilizumab and its long-term safety.
K e Y POINTS TO re M e MB er
AAION is characterized by a rapid onset of severe monocular vision loss with a relative afferent pupillary defect and pallid or hyperemic optic disc edema.
• AAION occurs because of inflammatory narrowing or occlusion of the posterior ciliary arteries by vasculitis (e.g., GCA).
• GCA is a granulomatous vasculitis that most commonly occurs in older white women.
• Systemic symptoms of GCA include temporal headache, jaw claudication, scalp tenderness, malaise, weight loss, and fever.
• About 50% of patients with GCA present with vision loss, in most cases due to AAION.
• High-dose corticosteroid treatment should be commenced immediately whenever GCA is suspected and should not be delayed while awaiting the results of laboratory studies or temporal artery biopsy.
Further Reading
González-Gay MA, García-Porrúa C, Llorca J, et al. Visual manifestations of giant cell arteritis: trends and clinical spectrum in 161 patients. Medicine. 2000;79:283–292.
Hayreh SS, Zimmerman B. Management of giant cell arteritis: our 27-year clinical study: new light on old controversies. Ophthalmologica. 2003;217:239–259.
Kawasaki A, Purvin V. Giant cell arteritis: an updated review. Acta Ophthalmol. 2009;87:13–32.
Parikh M, Miller NR, Lee AG, et al. Prevalence of a normal C-reactive protein with an elevated erythrocyte sedimentation rate in biopsy-proven giant cell arteritis. Ophthalmology. 2006;113:1842–1845.
Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis. N Engl J Med. 2017;377:317–328.