Abbreviations
4p-, 5p-, 22q- chromosome deletion syndromes for 4p, 5p, or 22q
AAP American Academy of Pediatrics
ACE angiotensin-converting enzyme
ADHD attention deficit hyperactivity disorder
AED antiepileptic drug
AFI amniotic fluid index
AFP α-fetoprotein
AIDS acquired immune deficiency syndrome
aOR adjusted odds ratio
AP anteroposterior
ARM anorectal malformation
ART assisted reproductive technology
ASD atrial septal defect
AV atrioventricular
AVM arteriovenous malformation
AVSD atrioventricular septal defect
BAV bicuspid aortic valve
BMI body mass index
BP breakpoint
BUN blood urea nitrogen
BW birth weight
Ca2+ serum calcium level
CBC complete blood count
CDC Centers for Disease Control and Prevention
CDH congenital diaphragmatic hernia
CDP chondrodysplasia punctata
cfDNA cell-free DNA (test)
CHD congenital heart disease or defects
Chr chromosome
CI confidence interval
CINCA chronic infantile neurological cutaneous articular
CK creatine kinase
CL cleft lip
CLO cleft lip only (without cleft palate)
CL/P cleft lip with or without cleft palate
CLP cleft lip with cleft palate
CMV cytomegalovirus
CNS central nervous system
CNV copy number variant
CoA coarctation of the aorta
CoQ10 coenzyme Q10 (ubiquinone)
CP cleft palate
CPAP continuous positive airway pressure
CPK creatine phosphokinase
CPO cleft palate only (without cleft lip)
CSF cerebrospinal fluid
CT computerized tomography
CTD conotruncal defect
CVS chorionic villous sampling
CVSD conoventricular septal defect
CSVT cerebral sinus venous thrombosis
DA/DC diamniotic dichorionic (twins)
DD developmental disability
DEB diepoxybutane
DES diethylstilbesterol
DEXA dual-energy X-ray absorptiometry
DI diabetes insipidus
DM diabetes mellitus
DNA deoxyribonucleic acid
DORV double-outlet right ventricle
DSD disorder of sex development
dTGA d-transposition of the great arteries
DVT deep venous thrombosis
DWM Dandy–Walker malformation
DZ dizygotic (twins)
ECG electrocardiogram
ECMO extracorporeal membrane oxygenation
EDTA ethylenediaminetetraacetic acid (lavender-top blood collection tube)
EEG electroencephalogram
ENT otolaryngology (ear–nose–throat)
EXIT ex utero intrapartum treatment
FDA US Food and Drug Administration
FISH fluorescence in situ hybridization
G tube gastrostomy tube
GA gestational age
GERD gastroesophageal reflux disease
GH growth hormone
GI gastrointestinal
GU genitourinary
HCG human chorionic gonadotropin
HCM hypertrophic cardiomyopathy
HIE hypoxic ischemic encephalopathy
HLHS hypoplastic left heart syndrome
HPE holoprosencephaly
HSV herpes simplex virus
IAAa, IAAb interrupted aortic arch, type a or b
IC imprinting center
ICP increased intracranial pressure
ICSI intracytoplasmic sperm injection
ID intellectual disability
IDM infant of a diabetic mother
IgA immunoglobulin A
IGF insulin-like growth factor
IgG immunoglobulin G
IgM immunoglobulin M
IQ intelligence quotient
IUFD intrauterine fetal demise
IUGR intrauterine growth retardation
IVC inferior vena cava
LBW low birth weight
LCHAD long-chain 3-hydroxyacyl-CoA dehydrogenase
LCMV lymphocytic choriomeningitis virus
LGA large for gestational age
LL lower limb
LVOTO left ventricular outflow tract obstruction
MAC microphthalmia/anophthalmia/coloboma
MC/DA monochorionic diamniotic (twins)
MC/MA monochorionic monoamniotic (twins)
MCV mean corpuscular volume
MIM Mendelian Inheritance in Man (database)
MLPA multiplex ligation-dependent probe amplification (test for duplications/deletions)
MMF mycophenolate mofetil
MMI methimazole
MoM multiple of the median
mPKU maternal phenylketonuria
MRA magnetic resonance angiography
MRI magnetic resonance imaging
MSAFP maternal serum α-fetoprotein
mtDNA mitochondrial DNA
mTOR mechanistic target of rapamycin (signaling pathway)
MZ monozygotic (twins)
NAI nonaccidental injury
NG nasogastric
NIH non-immune hydrops
NOMID neonatal-onset multisystem inflammatory disorder
NT nuchal translucency
NTD neural tube defect
OCA oculocutaneous albinism
OCD obsessive–compulsive disorder
OI osteogenesis imperfecta
OR odds ratio
OTIS Organization of Teratogen Information Services
PAE prenatal alcohol exposure
PAS perinatal arterial stroke
PCD primary ciliary dyskinesia
PCR polymerase chain reaction
PDA patent ductus arteriosus
PFO patent foramen ovale
Phe phenylalanine
PKU phenylketonuria
PPV positive predictive value
PS pulmonary stenosis
QT(c) distance between Q wave and T wave on ECG; QTc is QT interval corrected for heart rate
RNA ribonucleic acid
RPR rapid plasma reagin test (for syphilis)
RR relative risk
RSV respiratory syncytial virus
SA situs ambiguus
SCID severe combined immunodeficiency syndrome
SD standard deviation
SGA small for gestational age
SIDS sudden infant death syndrome
SLE systemic lupus erythematosus
SMA spinal muscular atrophy
SMMCI solitary median maxillary central incisor
SNHL sensorineural hearing loss
SNP single nucleotide polymorphism
SVT supraventricular tachycardia
T13, trisomy 13
T18 trisomy 18
T21 trisomy 21, Down syndrome
TA truncus arteriosus
TAPVR total anomalous pulmonary venous return
TE tracheoesophageal
TGA transposition of the great arteries
TOF tetralogy of Fallot
TPPA treponema pallidum particle agglutination assay
TS Turner syndrome
TSHR-SAb thyrotropin receptor-stimulating antibodies
TTTS twin-to-twin transfusion syndrome
uE3 unconjugated estriol 3
UL upper limb
UPD unipaternal disomy; maternal (matUPD) or paternal (patUPD)
US ultrasound
VCUG vesicourethrogram
VDRL Venereal Disease Research Laboratory (test for syphilis)
VLCAD very long-chain acyl-CoA dehydrogenase deficiency
VPA valproic acid
VSD ventricular septal defect
VZIG varicella zoster-specific immunoglobulin G
VZV varicella zoster virus
WES whole exome sequencing
WHO World Health Organization
WHS Wolf–Hirschhorn syndrome
1
Hypotonia
Clinical Consult
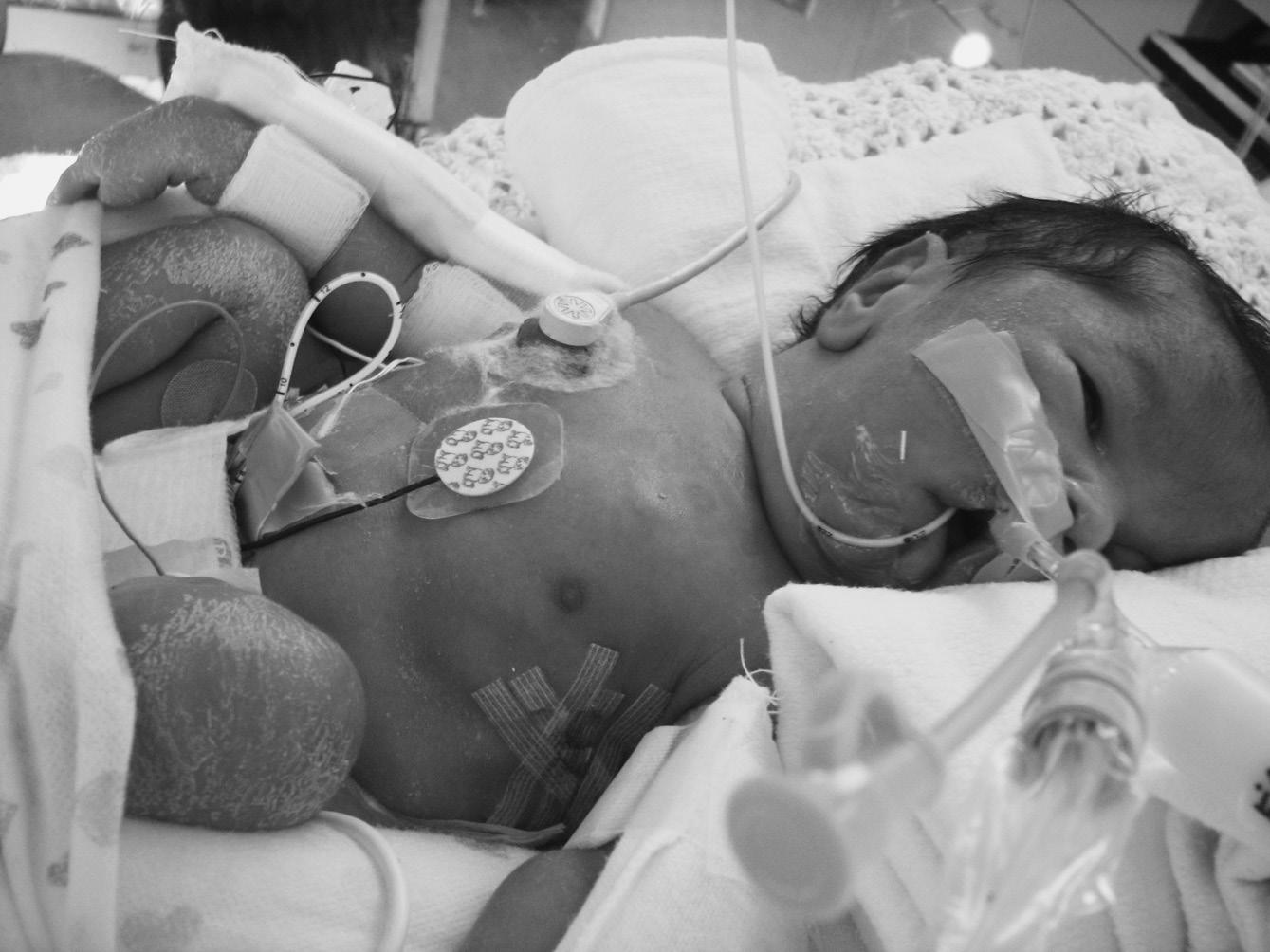
A term infant with hypotonia and bilateral metatarsus adductus presented with relative pulmonary hypoplasia requiring full ventilatory support. The mother had mild polyhydramnios and reported decreased fetal activity. The physical exam was challenging due to multiple tubes and monitoring devices. His mouth was tented (Figure 1.1). He had hypoactive reflexes and minimal spontaneous movements of arms, legs, and fingers. The initial diagnosis was a disorder with Fetal Akinesia sequence, and exome sequencing was considered.
The family history was pertinent for three healthy children. Another child died 2 years previously with presumptive hypoxic ischemic encephalopathy. An autopsy did not reveal an underlying cause. The parents had been given a low recurrence risk. The mother reported no history of weakness or difficulty releasing a grasped object. She had subtle facial weakness. No grasp myotonia was elicited on initial examinations by genetics and neurology consultants, but careful reassessment revealed weak eyelids and suggestive grasp myotonia. Molecular testing for myotonic dystrophy in the infant revealed an expansion of 1,200 CTG repeats in DMPK1, consistent with congenital myotonic dystrophy
Symptoms of myotonic dystrophy type 1 in affected mothers may be subtle and mild. Even experienced consultants can miss this diagnosis, which was the case when the first severely affected child was born to this mildly affected mother.
Definition
• Hypotonia is low muscle tone for age, often caused by weakness or abnormalities of the central nervous system (CNS).
• 2–4% of term infants
• Male = Female
• Major types of hypotonia
⚬ Central hypotonia ~80%
▪ Chromosome disorders (most frequent), CNS lesions, metabolic disorders
▪ Clinical features: reflexes present, variable seizures and dysmorphic features
⚬ Peripheral hypotonia ~20%
▪ Congenital myopathies, spinal muscular atrophy, myotonic dystrophy
▪ Clinical features: reflexes hypoactive, lack of antigravity movements
• Prenatal findings often associated with hypotonia
⚬ Decreased fetal movements
⚬ Polyhydramnios
⚬ Breech presentation
Differential Diagnosis
• We outline only a few of the hundreds of genetic conditions that can cause neonatal hypotonia. Many chromosomal and microarray abnormalities, single gene disorders, various metabolic diseases, and numerous complex syndromes cause congenital hypotonia. Multiple brain malformations are an important cause of neonatal hypotonia, but these are discussed in other chapters.
• The pace of new gene discoveries in infants with hypotonis is astonishing and as molecular pathways are elucidated, therapeutic targets are emerging.
• Using all tools available, a diagnosis can be achieved in ~90% of hypotonic infants. A detailed physical examination and thorough history remain essential for diagnosis, even in the genomic era.
⚬ More than 50% of patients can be diagnosed by exam and history alone.
⚬ In the remaining patients, a well-defined clinical phenotype facilitates choice of diagnostic tests and interpretation of molecular results.
• Chromosome disorders
⚬ Down syndrome* (MIM 190685)
▪ Clinical features: characteristic facial findings, small ears < 3%, theatrical grimace (when crying), short broad hands with fifth finger clinodactyly, sandal gap, congenital heart defects in almost half
⚬ Prader–Willi syndrome* (MIM 176270)
▪ Missing or inactive paternal contribution at chr 15q11.2
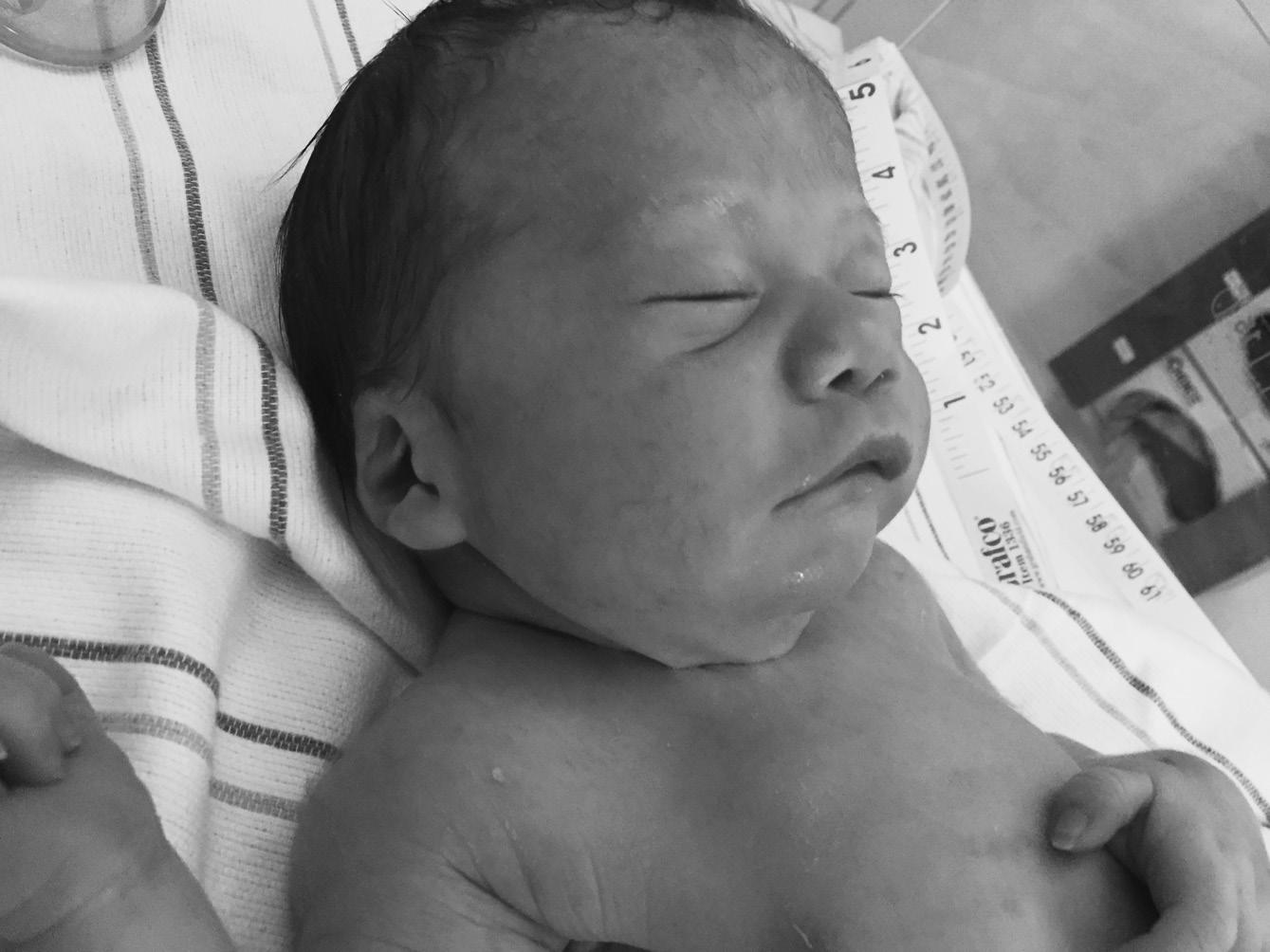
▪ Clinical features: congenital generalized hypotonia a constant feature (Figure 1.2), poor feeding, absent or reduced suck, frog leg positioning, reflexes present, genital hypoplasia, clitoral hypoplasia (often overlooked in females—a helpful sign), cryptorchidism, decreased pigmentation for family background
Pearl: Unexplained poor feeding in a term infant without congenital anomalies warrants a brain MRI. If the MRI is normal, follow with DNA methylation study to rule out Prader–Willi syndrome.
⚬ Smith–Magenis syndrome (MIM 182290)
▪ Deletion of 3.7-Mb interstitial deletion in chromosome 17p11.2
• 10% caused by mutation in the RAI1 gene at 17p11.2
FIGURE 1.1 Congenital myotonic dystrophy in a neonate with pulmonary hypoplasia, tented mouth, facial edema, and hypotonic posture due to
FIGURE 1.2 Hypotonia in baby with Prader–Willi syndrome. Note frog leg position of comfort and typical facial features.
FIGURE 1.3 Smith–Magenis syndrome in an infant with dysmorphic features including small ears, downturned mouth corners, and lower canthal folds Note copious oral secretions reflecting decreased oral–motor tone.
▪ Clinical features: small ears, brachycephaly, midface hypoplasia, prognathism, hoarse cry, cardiac and other defects, seizures, intellectual disability (ID) (Figure 1.3)
▪ Later: sleep problems and characteristic behaviors
⚬ 12p tetrasomy (Pallister–Killian mosaicism, MIM 601803)
▪ Mosaic marker chromosome consisting of two copies of short arm of chr 12; not present in all tissues
▪ Clinical features: congenital diaphragmatic hernia, small ears, bitemporal alopecia, characteristic face and upper lip (see Figure 3.2), feeding problems, seizures, severe ID
⚬ MECP2 duplication at Xq28 (MIM 300260)
▪ Variable, mostly small <1-Mb duplications on chr Xq28, diagnosed on chromosome single nucleotide polymorphism (SNP) microarray; usually diagnosed in males
▪ Clinical features: severe ID, hypotonia and spasticity, recurrent respiratory infections, neonatal renal calculi
• Dysmorphic single gene syndromes
⚬ Kabuki syndrome (MIM 147920)
▪ Autosomal dominant disorder, caused by heterozygous variants in MLL2 (KMT2D)
▪ Clinical features: cleft palate, cardiac defects, genitourinary defects, mildly myopathic face with long palpebral fissures, blue sclerae, everted lateral third of lower eyelids, prominent fingertip pads, short fifth fingers (see Figure )
⚬ Smith–Lemli–Opitz syndrome* (MIM 270400)
▪ Autosomal recessive disorder of cholesterol metabolism caused by variants in DCHR7
▪ A multiple congenital anomaly syndrome with cleft palate, polydactyly, genital ambiguity, cardiac anomalies, and intrauterine growth restriction. Characteristic face with ptosis and anteverted nares.
▪ Hypotonia a constant finding
⚬ Rett syndrome variant (MIM 613454)
▪ Autosomal dominant, caused by de novo heterozygous variant in FOXG1
• Occasionally, a parent has germline mosaicism.
▪ Clinical features: progressive microcephaly, developmental delay, ID, stereotypic movements of hands
• Hypoplasia of corpus callosum
⚬ Bohring–Opitz syndrome (MIM 605039)
▪ Autosomal dominant, caused by de novo heterozygous variants in AXL1
▪ Clinical findings: distinctive facial features, variable microcephaly, nevus flammeus, hypertrichosis, severe myopia, unusual posture of arms with flexion at elbows and wrists, hypotonia, severe feeding problems with vomiting, severe ID
▪ Increased risk for Wilms tumor; ultrasound (US) surveillance indicated
⚬ PURA associated hypotonia witn neonatal respiratory distress (MIM 616158)
▪ Autosomal dominant , usually de novo variants in PURA
▪ Gene responsible for much of phenotype seen in 5q31.3 deletion syndrome
▪ Broad prominent forehead; myopathic face
▪ May mimic HIE as neonatal apnea and seizures common; non-progressive but severe ID.
• Metabolic disorders
⚬ Zellweger syndrome (peroxisome biogenesis disorder 1A, MIM 214100)
▪ Lethal autosomal recessive disorder, caused by homozygous or compound heterozygous variants in PEX1 and other peroxisome biogenesis genes
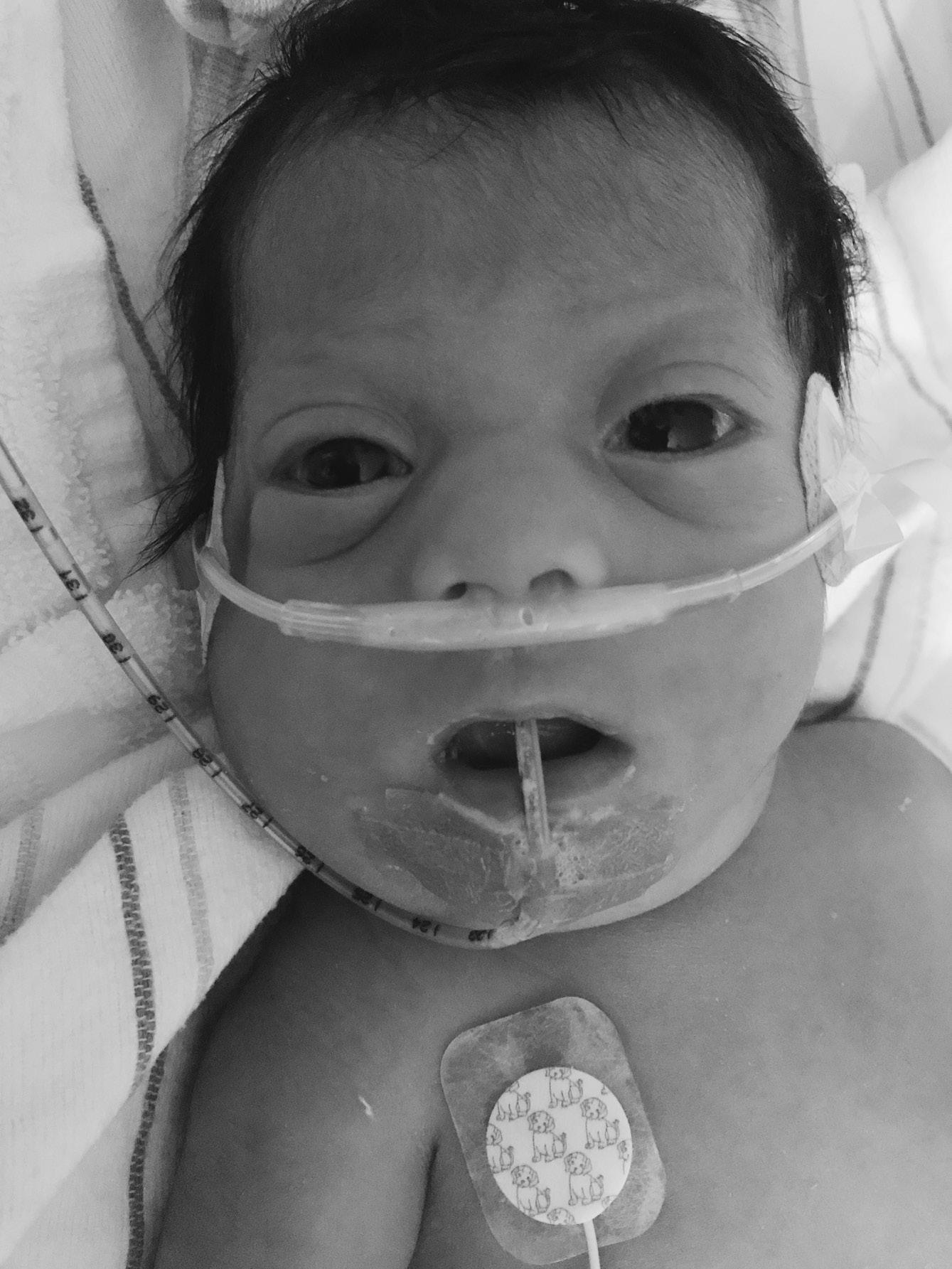
▪ Clinical features: characteristic face with high forehead, large fontanelles (Figure 1.4), severe progressive hypotonia, hepatomegaly, liver disease, poor feeding and seizures
⚬ Congenital disorders of glycosylation (CDG-1A, MIM 212605)
▪ Expanding group of autosomal recessive disorders of protein glycosylation
• CDG-1A is the most common type, caused by biallelic variants in PMM2.
▪ Two main groups based on type of biochemical error: type I CDG and type II CDG
▪ Clinical features: highly variable: liver disease, failure to thrive, microcephaly, developmental delay, dysmorphic features, abnormal fat distribution on buttocks and elsewhere
• Brain MRI: cerebellar hypoplasia and other CNS lesions
▪ Labs: carbohydrate-deficient transferrin electrophoresis and/or peroxisomal disorders sequencing panel
⚬ Glycine encephalopathy (nonketotic hyperglycinemia, MIM 605899)
▪ Autosomal recessive disorder, caused by variants in GLCD, which encodes the P protein (MIM 238300) in
the mitochondrial glycine cleavage system. Other genes encode the T protein, GCST (MIM 238310); rarely others.
• The neonatal form of nonketotic hyperglycinemia is apparent in the first few days after birth.
▪ Clinical features: encephalopathy, failure to thrive, lethargy, severe illness, intractable seizures
• Hiccups are a frequent and helpful clue.
• Brain imaging: hydrocephalus, mega cisterna magna, white matter atrophy, corpus callosum hypoplasia
▪ Labs: Glycine accumulates in blood, urine, and cerebrospinal fluid.
• It may not be detected by newborn screening programs.
▪ Treat with sodium benzoate and dextromethorphan. Neonatal death is frequent, and survivors have severe ID.
⚬ FBXL4-related encephalomyopathic mitochondrial DNA (mtDNA depletion syndrome, MIM 605471)
▪ Autosomal recessive disorder, due to biallelic variants in FBXL4
▪ Clinical features: congenital or early onset lactic acidosis, growth failure, feeding difficulty, hypotonia, global delay, seizures, movement disorders, ataxia, autonomic dysfunction, stroke-like episodes
• Hypertrophic cardiomyopathy, congenital heart malformations, arrhythmias
• Cataract, strabismus, nystagmus, optic atrophy
• Brain MRI: basal ganglia changes, periventricular cysts, cerebellar abnormalities, thin corpus callosum
▪ Labs: elevated transaminases, lactic acidosis, neutropenia, hyperammonemia, mild creatine kinase (CK) elevations
▪ Early death; median age at death, 2 years
⚬ Primary coenzyme Q10 (CoQ10 deficiency type I, MIM 607426)
▪ Autosomal recessive disorder, caused by biallelic variants in nine genes involved in synthesis of CoQ
▪ Clinical features: multisystem disease that may present as fatal neonatal encephalopathy with hypotonia
• Steroid-resistant nephrotic syndrome may be initial manifestation. This occasionally may be an isolated finding.
• Hypertrophic cardiomyopathy, retinopathy, optic atrophy, hearing loss
• Later onset forms: slowly progressive multiple system disorder with parkinsonism, cerebellar ataxia, pyramidal dysfunction, dystonia, spasticity, seizures, ID
▪ Labs: biochemical demonstration on frozen muscle homogenates of reduced levels of CoQ10 (ubiquinone) in skeletal muscle or of complex I + III and II + III of the mitochondrial respiratory chain
▪ Treat with oral high-dose CoQ10
• Neonatal death is frequent. There is occasional long survival with late-onset renal disease and neurologic and autonomic symptoms.
Pearl: Serum coenzyme Q levels reflect dietary intake. CoQ10 levels in a tissue biopsy (preferably skeletal muscle) are needed to make the diagnosis of deficiency.
⚬ Other metabolic disorders
▪ Most are autosomal recessive and many are detected on newborn screening.
• Rare inborn errors of metabolism are identified with exome sequencing or targeted panels.
▪ Aminoacidurias
• Methylmalonic aciduria (MIM 251000)
• Maple syrup urine disease (MIM 248600)
• Propionic acidemia (MIM 606054)
▪ Lysosomal storage diseases
• Pompe disease (glycogen storage disease type II, MIM 232300)
• Congenital lower motor neuron diseases
⚬ Spinal muscular atrophy I (Werdnig–Hoffman disease, SMA1, MIM 255300)
▪ Autosomal recessive disorder: >95% of patients have a homozygous deletion in SMN1 on chromosome 5q.
FIGURE 1.4 Infant with Zellweger syndrome. Note tented mouth, hypertelorism, and high forehead.
▪ Clinical features: alert infant, joint contractures, absent reflexes, variable tongue fasciculations
• Relentless, progressive weakness due to lower motor neuron dysfunction, with eventual respiratory failure
▪ Treatment
• Multiple clinical trials are in progress to ameliorate disease process; see https://clinicaltrials.gov.
• Spinraza (Nusinersen) was approved by the U.S. Food and Drug Administration (FDA) in late 2016. Clinical trials suggest that motor milestones may be maintained when it is given to presymptomatic infants with later infancy onset forms of spinal muscular atrophy.
⚬ Spinal muscular atrophy with respiratory distress (SMARD1, MIM 604320)
▪ Autosomal recessive disorder, caused by biallelic variants in IGHMBP2
▪ Clinical features: early weakness predominantly involving distal muscles, upper limb > lower limb, diaphragmatic eventration, respiratory infections, mild knee and foot contractures
• Diaphragmatic palsy requires supportive ventilation.
• Autonomic involvement: pain insensitivity, bowel and bladder problems, excessive sweating
▪ Usually lethal during infancy
• Congenital muscular dystrophies and myopathies
⚬ Clinically and genetically heterogeneous group of disorders, with inconsistent and complex terminology and classification systems. Pathogenic variants in the same gene can produce variable phenotypes in both the myopathy and muscular dystrophy categories.
⚬ Collagen VI-related dystrophies
▪ Ullrich congenital muscular dystrophy (MIM 254090)
• Autosomal recessive trait, caused by biallelic mutations in one of three collagen VI genes: COL6A1, COL6A2, COL6A3
• Clinical features: most severe end of spectrum has striking joint hypermobility of hands and feet, congenital hip dislocation, clubfeet, elbow and knee contractures, kyphoscoliosis, torticollis, progressive weakness
• Respiratory insufficiency, especially at night
• Normal IQ
▪ Bethlem myopathy (MIM 158810)
• Autosomal dominant trait, allelic to Ullrich congenital muscular dystrophy but milder, caused by heterozygous mutations in same three collagen VI genes
Pearl: A baby with both congenital hypotonia or joint laxity can present with contractures (e.g., SMA1, Marfan syndrome, Loeys–Dietz syndrome, and Ullrich muscular dystrophy).
⚬ Congenital myotonic dystrophy type 1 (MIM 160900) (see Clinical Consult)
▪ Autosomal dominant disorder, caused by expanded number of trinucleotide (CTG) repeats in DMPK on chromosome 19p
• Congenital presentation typically has >1,000 CTG repeats and is inherited from an affected mother, who may be mildly affected or asymptomatic.
▪ Prenatal findings: polyhydramnios, decreased fetal activity, hydrops of upper body
▪ Clinical features: variable severity, myopathic face, tented triangular mouth, high forehead, clubfeet; hypoactive reflexes, significant feeding and swallowing problems, oromotor weakness/dysfunction
• Hypotonia and respiratory status usually improve gradually, but severe pulmonary hypoplasia may cause neonatal death.
• There is global but variable developmental delay.
▪ Maternal findings: subtle craniofacial weakness, mild bitemporal hair loss, grasp myotonia, learning disability, unconcerned affect (“la belle indifference”)
• Affected mothers often deny symptoms. Ask about grasp myotonia in several ways: “Do you ever have difficulty letting go of a hairbrush or the steering wheel?” and “Do your hands ever get ‘stuck’ after picking up a heavy object?”
Pearl: The electromyogram (EMG), diagnostic of myotonic dystrophy in older children and adults, is usually not helpful in neonates.
⚬ Laminin alpha 2-related dystrophy (LAMA2-RD, congenital merosin-deficient muscular dystrophy, MIM156225)
▪ Autosomal recessive disorder, caused by biallelic variants in LAMA2
• Complete LAMA2 deficiency causes approximately half of congenital muscular dystrophy.
• Partial deficiency has a more variable course.
▪ Clinical features: hypotonia, weakness and occasional joint contractures; seizures 30%; several other presentations
• Brain MRI: subcortical cysts, white matter changes, subcortical band heterotopia in 5%, variable cerebellar and pontine changes
• Labs: CK elevated up to five times normal
⚬ Muscular dystrophy with eye and brain anomalies (dystroglycanopathies, Walker–Warburg syndrome, muscle–eye–brain disease, MIM, 613150, 236670 and others)
▪ Autosomal recessive disorders, caused by bialleic variants in POMT1, POMT2, FKTN, and others
• A genetically heterogeneous group of disorders resulting from defective glycosylation of dystrophinassociated glycoprotein 1
▪ Clinical features: macrocephaly or microcephaly, cleft lip/palate, contractures
• Eye: retinal malformations, microphthalmia, cataract, glaucoma, anterior chamber abnormalities
• Brain MRI: a range from cobblestone (type II) lissencephaly to more focal polymicrogyria; cerebellar malformations, hypoplasia of midline brain structures, ventricular dilatation, Dandy–Walker malformation, posterior occipital encephalocele
▪ Labs: strikingly elevated CK
▪ Variable course from neonatal lethal to death in the first year, to less severe presentations
⚬ RYR-related myopathies (central core disease, minicore myopathy, MIM 180901)
▪ Autosomal recessive disorder, caused by biallelic variants in RYR1, which encodes the sarcoplasmic reticulum calcium release channel
• Recessive pathogenic variants usually result in a neonatal presentation.
▪ Clinical features: facial weakness, early onset severe progressive scoliosis
• Ophthalmoplegia in some forms of RYR1 myopathy (central core and minicore myopathy) but may be absent in the congenital presentation
▪ May require gastrostomy and nighttime ventilator support
• Nongenetic causes:
⚬ Most are not addressed here, including prematurity, congenital infection, botulism, hypothyroidism, cardiac failure, anemia, hypoxic and hemorrhagic brain lesions, hypothyroidism, and perinatal stroke. Hypoxic ischemic encephalophathy is discussed briefly.
⚬ Hypoxic ischemic encephalopathy (HIE)
▪ HIE is caused by acute or chronic in utero or perinatal asphyxia.
▪ Incidence 1–2/1,000 live births
▪ Common clinical features: cord blood pH <7.0; base excess >12; Apgar score <3 for greater than 5 minutes; early onset moderate or severe encephalopathy; seizures within 12 hours of life, unresponsive to pyridoxine; multi- organ dysfunction: liver, kidney, lung
• Magnetic resonance imaging (MRI)/computerized tomography (CT) show characteristic findings consistent with asphyxia; early brain MRI helps date injury
• Other disease processes must be excluded.
Pearl: Low Apgar scores are neither necessary nor sufficient to make a diagnosis of HIE. Many other conditions cause low Apgar scores.
Evaluation and Management
• Review pregnancy history: polyhydramnios, commonly due to reduced fetal swallowing, abnormal fetal lie, and decreased fetal activity.
⚬ Document fetal ultrasound abnormalities (e.g., clubfeet).
⚬ Note delivery complications, Apgar scores, resuscitation status at birth, and cord gases.
• Document the family history: consanguinity, infant death, weakness.
• Examine the parents for weakness and, especially in the mother, for slow grip release and other findings of myotonic dystrophy.
• Examine for distinctive features. Careful phenotyping will increase diagnostic yield.
⚬ Dysmorphic features: e.g. cleft palate, micrognathia, large fontanelles (e.g. Zellweger syndrome)
⚬ Eye exam for retinal dystrophy: e.g. in muscular dystrophy with eye and brain anomalies (Walker–Warburg) syndrome
⚬ Genitalia: e.g. small phallus, clitoral hypoplasia (e.g. Prader–Willi syndrome*), cryptorchidism, genital ambiguity (e.g. Smith–Lemli–Opitz syndrome*)
⚬ Neurological exam: Consult pediatric neurology to refine the neurologic phenotype.
▪ Reflexes (absent in spinal muscular atrophy), extra ocular movements, seizures, head control, spontaneous movement of extremities, muscle bulk and so on.
• Imaging
⚬ Skeletal survey for epiphyseal stippling in suspected peroxisomal disorders
▪ Order lateral view of the foot for heel stippling.
⚬ Abdominal US for organomegaly
⚬ Echocardiogram for cardiomegaly (Pompe disease MIM 232300)
⚬ Neuroimaging establishes the diagnosis in ~25%.
▪ MRI gives best definition of anatomy.
▪ Brain CT scan is useful for hemorrhagic events.
• Genetic testing
⚬ Chromosome analysis (for suspected trisomy 21); SNP microarray in all others. Excessive homozygosity may direct further testing.
⚬ DNA methylation for chr 15q11.2 for suspected Prader–Willi syndrome*
⚬ Metabolic studies for hypotonia without an identified cause
▪ Use an experienced reference laboratory.
▪ Review newborn screening test results.
▪ Initial baseline tests: quantitative urine organic acids, quantitative plasma amino acids, plasma and urine carnitine (total and esterified), plasma acylcarnitine profile, serum lactate, CK, ammonia, T4, thyroidstimulating hormone
Pearl: Elevated CK narrows the differential diagnosis. Include it in initial round of tests.
⚬ For suspected congenital disorders of glycosylation
▪ Serum carbohydrate-deficient transferrin analysis and plasma N-glycan profile
⚬ For suspected peroxisomal disorders, begin with biochemical testing.
▪ Plasma very long-chain fatty acids (VLCFAs)
• Elevated C26:0 and C26:1 and C24/C22 and C26/ C22 ratios
• When VLCFA is abnormal, consult with reference laboratory for further testing.
• Consider peroxisomal disorders sequencing panel to expedite diagnosis
• Patients with mild Zellweger syndrome may have (near) normal biochemical tests in plasma and urine, and further tests in fibroblasts or a gene panel may be needed.
⚬ Molecular genetic testing should be guided by clinical findings.
⚬ Single gene tests
▪ Spinal muscular atrophy: exonic deletion in SMN1
▪ Congenital myotonic dystrophy: increased CTG trinucleotide repeats in DMPK
⚬ Gene panels or exome sequencing trio testing (child and both parents if possible) for complex or unusual phenotypes
▪ Consult genetic experts to interpret results and variants of unknown significance.
Pearl: Trinucleotide repeat disorders such as congenital myotonic dystrophy cannot be diagnosed with exome sequencing.
⚬ Locate genetic laboratories at
▪ https://www.genetests.org
▪ https://www.ncbi.nlm.nih.gov/gtr
⚬ Reserve invasive testing (EMG, nerve conduction, and muscle biopsy) for infrequent select cases.
▪ Additional tests on muscle biopsy may include immunohistochemistry staining, electron
microscopy, and respiratory chain enzyme analysis of mitochondrial DNA.
▪ Include CoQ10 levels in skeletal muscle homogenates.
▪ Increasingly, molecular testing (panels, exomes) is replacing invasive testing.
Further Reading
Bushby KM, Collins J, Hicks D. (2014) Collagen type VI myopathies. Adv Exp Med Biol. 802:185–99. PMID 24443028
Falsaperla R, Praticò AD, Ruggieri M, et al. (2016) Congenital muscular dystrophy: from muscle to brain. Ital J Pediatr. 42(1):78. PMID 27576556
Gonorazky HD, Bönnemann CG, Dowling JJ. (2018) The genetics of congenital myopathies. Handb Clin Neurol.148:549–564. PMID 29478600
Groen EJN, Talbot K, Gillingwater TH. (2018) Advances in therapy for spinal muscular atrophy: promises and challenges. Nat Rev Neurol. 14(4):214–24. PMID 29422644
Jungbluth H, Ochala J, Treves S, Gautel M. (2017) Current and future therapeutic approaches to the congenital myopathies. Semin Cell Dev Biol. 64:191–200. PMID 27515125
Salviati L, Trevisson E, Doimo M, Navas P. (2017) Primary coenzyme Q10 deficiency. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2017. PMID 28125198
Sparks SE, Krasnewich DM. (2017) Congenital disorders of N-linked glycosylation and multiple pathway overview. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2017. PMID 20301507
Tanaka AJ, Bai R, Cho MT et al. (2015) De novo mutations in PURA are associated with hypotonia and developmental delay. Cold Spring Harb Mol Case Stud. E pub PMID 27148565
Tarailo-Graovac M, Wasserman WW, Van Karnebeek CD. (2017) Impact of next-generation sequencing on diagnosis and management of neurometabolic disorders: Current advances and future perspectives Expert Rev Mol Diagn. 17:307–9. PMID 28277145